
Levonantradol: Difference between revisions
m Bot: Migrating 3 interwiki links, now provided by Wikidata on d:q2073800 (Report Errors) |
Fix. |
||
| (52 intermediate revisions by 29 users not shown) | |||
| Line 1: | Line 1: | ||
{{Short description|Chemical compound}} |
|||
{{Drugbox |
|||
{{Infobox drug |
|||
| verifiedrevid = 447558276 |
| verifiedrevid = 447558276 |
||
| IUPAC_name = [(6S,6aR,9R,10aR)- 9-hydroxy- 6-methyl- 3-[(2R)-5-phenylpentan- 2-yl]oxy- 5,6,6a,7,8,9,10,10a-octahydrophenanthridin- 1-yl] acetate |
| IUPAC_name = [(6S,6aR,9R,10aR)- 9-hydroxy- 6-methyl- 3-[(2R)-5-phenylpentan- 2-yl]oxy- 5,6,6a,7,8,9,10,10a-octahydrophenanthridin- 1-yl] acetate |
||
| Line 6: | Line 7: | ||
<!--Clinical data--> |
<!--Clinical data--> |
||
| tradename = |
| tradename = |
||
| legal_status = |
| legal_status = |
||
| routes_of_administration = |
| routes_of_administration = |
||
<!--Pharmacokinetic data--> |
<!--Pharmacokinetic data--> |
||
| metabolism = |
| metabolism = |
||
| elimination_half-life = |
| elimination_half-life = |
||
| excretion = |
| excretion = |
||
<!--Identifiers--> |
<!--Identifiers--> |
||
| Line 27: | Line 28: | ||
<!--Chemical data--> |
<!--Chemical data--> |
||
| C=27 | H=35 | N=1 | O=4 |
| C=27 | H=35 | N=1 | O=4 |
||
| molecular_weight = 437.571 g/mol |
|||
| smiles = O=C(Oc2cc(O[C@H](C)CCCc1ccccc1)cc4c2[C@@H]3C[C@H](O)CC[C@H]3[C@@H](N4)C)C |
| smiles = O=C(Oc2cc(O[C@H](C)CCCc1ccccc1)cc4c2[C@@H]3C[C@H](O)CC[C@H]3[C@@H](N4)C)C |
||
| InChI = 1/C27H35NO4/c1-17(8-7-11-20-9-5-4-6-10-20)31-22-15-25-27(26(16-22)32-19(3)29)24-14-21(30)12-13-23(24)18(2)28-25/h4-6,9-10,15-18,21,23-24,28,30H,7-8,11-14H2,1-3H3/t17-,18+,21-,23+,24-/m1/s1 |
|||
| InChIKey = FFVXQGMUHIJQAO-BFKQJKLPBR |
|||
| StdInChI_Ref = {{stdinchicite|correct|chemspider}} |
| StdInChI_Ref = {{stdinchicite|correct|chemspider}} |
||
| StdInChI = 1S/C27H35NO4/c1-17(8-7-11-20-9-5-4-6-10-20)31-22-15-25-27(26(16-22)32-19(3)29)24-14-21(30)12-13-23(24)18(2)28-25/h4-6,9-10,15-18,21,23-24,28,30H,7-8,11-14H2,1-3H3/t17-,18+,21-,23+,24-/m1/s1 |
| StdInChI = 1S/C27H35NO4/c1-17(8-7-11-20-9-5-4-6-10-20)31-22-15-25-27(26(16-22)32-19(3)29)24-14-21(30)12-13-23(24)18(2)28-25/h4-6,9-10,15-18,21,23-24,28,30H,7-8,11-14H2,1-3H3/t17-,18+,21-,23+,24-/m1/s1 |
||
| Line 37: | Line 35: | ||
| StdInChIKey = FFVXQGMUHIJQAO-BFKQJKLPSA-N |
| StdInChIKey = FFVXQGMUHIJQAO-BFKQJKLPSA-N |
||
}} |
}} |
||
| ⚫ | '''Levonantradol''' ('''CP 50,556-1''') is a [[Chemical synthesis|synthetic]] [[cannabinoid]] [[Analog (chemistry)|analog]] of [[dronabinol]] (Marinol) developed by [[Pfizer]] in the 1980s. It is around 30 times more potent than [[THC]], and exhibits [[antiemetic]] and [[analgesic]] effects via activation of CB<sub>1</sub> and CB<sub>2</sub> [[cannabinoid receptor]]s.<ref>{{cite journal | vauthors = Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR | title = Pharmacology and stereoselectivity of structurally novel cannabinoids in mice | journal = The Journal of Pharmacology and Experimental Therapeutics | volume = 247 | issue = 3 | pages = 1046–1051 | date = December 1988 | pmid = 2849657 }}</ref> Levonantradol is not currently used in medicine as [[dronabinol]] or [[nabilone]] are felt to be more useful for most conditions, however it is widely used in research into the potential therapeutic applications of cannabinoids.<ref>{{cite journal | vauthors = Tramèr MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ | title = Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review | journal = BMJ | volume = 323 | issue = 7303 | pages = 16–21 | date = July 2001 | pmid = 11440936 | pmc = 34325 | doi = 10.1136/bmj.323.7303.16 }}</ref><ref>{{cite journal | vauthors = Campbell FA, Tramèr MR, Carroll D, Reynolds DJ, Moore RA, McQuay HJ | title = Are cannabinoids an effective and safe treatment option in the management of pain? A qualitative systematic review | journal = BMJ | volume = 323 | issue = 7303 | pages = 13–16 | date = July 2001 | pmid = 11440935 | pmc = 34324 | doi = 10.1136/bmj.323.7303.13 }}</ref><ref>{{cite journal | vauthors = Ben Amar M | title = Cannabinoids in medicine: A review of their therapeutic potential | journal = Journal of Ethnopharmacology | volume = 105 | issue = 1–2 | pages = 1–25 | date = April 2006 | pmid = 16540272 | doi = 10.1016/j.jep.2006.02.001 | citeseerx = 10.1.1.180.308 }}</ref> |
||
| ⚫ | |||
| ⚫ | '''Levonantradol''' ('''CP 50,556-1''') is a [[Chemical synthesis|synthetic]] [[cannabinoid]] [[Analog (chemistry)|analog]] of [[dronabinol]] (Marinol) developed by [[Pfizer]] in the 1980s. It is around |
||
| ⚫ | Levonantradol is a full CB<sub>1</sub> receptor agonist. Cannabinoid receptors belong to the superfamily of [[G-protein coupled receptors]] (GPCRs), and endogenous cannabinoids naturally activate GPCRs. GPCRs modulate the inhibition of [[adenylyl cyclase]] and accumulation of the second messenger, [[cyclic adenosine monophosphate]] (cAMP). The CB<sub>1</sub> receptor is the most common GPCR in the central nervous system. The activation of CB<sub>1</sub>Rs decrease calcium conductance and increase potassium conductance in the brain. CB signaling naturally modulates synaptic transmission and mediates psychoactivity, and synthetic cannabinoids mimic these same actions. Although the efficacy of Levonantradol is dependent on the level of GCPR activity, Full agonists like Levonantradol have the ability to activate GPCRs and convert G<sub>α</sub> into a high affinity state for GTP or low affinity state for GDP. Previous studies suggest that Levonantradol has a higher binding affinity and efficacy than other similar synthetic cannabinoids (e.g. Δ<sup>9</sup>-THC). |
||
| ⚫ | |||
| ⚫ | |||
| ⚫ | Although Levonantradol has been extensively tested on animals including cats, rodents, and non-human primates. It has also been tested among cancer patient populations in clinical trials. Levonantradol is most commonly administered intramuscularly (I.M.), however it can also be administered orally. The dosage can range from 0.25 mg-3.0 mg every 2–4 hours, and the half-life is 1–2 hours. In order to administer Levonantradol intramuscularly, the drug must be dissolved in 5% ethanol, 5% emulphur, and 90% sterile saline. Synthetic cannabinoids like Levonantradol readily cross the [[blood–brain barrier]] because they are highly lipophilic and have low molecular weights. Levonantradol's bioavailability is variable due to the first pass metabolism. |
||
| ⚫ | |||
| ⚫ | Levonantradol is a full CB<sub>1</sub> receptor agonist. Cannabinoid receptors belong to the superfamily of [[G-protein coupled receptors]] (GPCRs), and endogenous cannabinoids naturally activate GPCRs. GPCRs modulate the inhibition of [[adenylyl cyclase]] and accumulation of the second messenger, [[cyclic adenosine monophosphate]] (cAMP). The CB<sub>1</sub> receptor is the most common GPCR in the central nervous system. The activation of CB<sub>1</sub>Rs decrease calcium conductance and increase potassium conductance in the brain. CB signaling naturally modulates synaptic transmission and mediates psychoactivity, and synthetic cannabinoids mimic these same actions. Although the efficacy of Levonantradol is dependent on the level of GCPR activity, Full agonists like Levonantradol have the ability to activate GPCRs and convert G<sub>α</sub> into a high affinity state for GTP or low affinity state for GDP. Previous studies suggest that Levonantradol has a higher binding affinity and efficacy than other similar synthetic cannabinoids (e.g. Δ<sup>9</sup>-THC). |
||
| ⚫ | Levonantradol has been clinically tested in cancer patients for its pain relief and antiemetic benefits. Cancer patients that endure chemotherapy often develop intense nausea, and Levonantradol has been tested to reduce these emetic symptoms. It is often used instead of THC because it has a higher efficacy. Levonantradol also acts on pain pathways in the central nervous system, which enables the drug to alleviate pain. Studies have shown an absence of emetic side effects within the half-life of the Levonantradol administered. Other studies suggest that cannabinoid agonists can synergize opioid anti-nociception. Cannabinoid receptors are located in [[nociceptive]] pathways, and CBs can promote signal transduction in TRP channels. Although Levonantradol relieves nociceptive and postoperative pain, decreases nausea, and improves spasticity in addition to being more effective than placebos, it has yet to be approved as legal medicine. Researchers have concluded that Levonantradol is no more effective than [[Codeine]], which is why they do not recommend expansion into clinical practice. |
||
==Side effects== |
|||
| ⚫ | |||
| ⚫ | The side effects for Levonantradol include ptosis, sedation, and ataxia in non-human primates. In rodents, the symptoms include dysphoria, memory impairment, motor incoordination, reduced concentration, and disorientation. Levonantradol also decreases startle response. In humans, side effects include dry mouth, drowsiness, dizziness, altered perception, mild sedation, and lack of concentration. It can cause an increase in heart rate and decrease in blood pressure. Euphoric symptoms rarely occurred in subjects. |
||
==Synthesis== |
|||
| ⚫ | Although Levonantradol has been extensively tested on animals including cats, rodents, and non-human primates. It has also been tested among cancer patient populations in clinical trials. Levonantradol is most commonly administered intramuscularly (I.M.), however it can also be administered orally. The dosage can range from 0.25 |
||
[[File:Nantradol synthesis.svg|thumb|center|700px|Nantradol synthesis:<ref>{{cite journal|doi=10.1002/jhet.5570170841|title=Recent discoveries in the search for non-opiate analgetics|journal=Journal of Heterocyclic Chemistry|volume=17|issue=8|pages=1817–1820|year=1980| vauthors = Johnson MR, Milne GM }}</ref> asymmetric:<ref>{{cite journal | vauthors = Sheshenev AE, Boltukhina EV, Hii KK | title = Levonantradol: asymmetric synthesis and structural analysis | journal = Chemical Communications | volume = 49 | issue = 35 | pages = 3685–3687 | date = May 2013 | pmid = 23535893 | doi = 10.1039/C3CC41388H | authorlink3 = Mimi Hii }}</ref>]] |
|||
[[Dane salt]] formation between 3,5-dimethoxyaniline and [[ethyl acetoacetate]] followed by borohydrate reduction gives [[synthon]] '''1'''. The amino group is protected by rxn with [[ethyl chloroformate]], the ester group is saponified, and then cyclodehydration with [[polyphosphoric acid]] leads to the dihydroquinoline ring system ('''2'''). Deblocking with HBr is followed by etherification of the nonchelated phenolic hydroxyl gives '''3'''. Treatment with NaH and [[ethyl formate]] results in both N-formylation and C-formylation of the active methylene to give '''4'''. [[Michael addition]] of [[methyl vinyl ketone]] (MVP) followed by successive base treatments to remove the activating C-formyl group and then to complete the [[Robinson annulation]] to give '''5'''. Lithium in liquid ammonia reduces the olefinic linkage and successive acetylation and sodium borohydrate reductions complete the synthesis of nantradol ('''6'''). |
|||
==Related compounds== |
|||
| ⚫ | |||
Numerous other compounds similar to levonantradol were also developed at the same time, including [[CP 42,096]], [[CP 47,497]], [[CP 55,940]] and [[CP 55,244]]. The desacetyl derivative of levonantradol (DALN or CP 54,939) and its ''N''-methyl derivative, as well as the tetracyclic analogue all have similar activity to levonantradol itself.<ref>{{cite journal | vauthors = Howlett AC, Johnson MR, Melvin LS, Milne GM | title = Nonclassical cannabinoid analgetics inhibit adenylate cyclase: development of a cannabinoid receptor model | journal = Molecular Pharmacology | volume = 33 | issue = 3 | pages = 297–302 | date = March 1988 | pmid = 3352594 }}</ref> |
|||
:[[Image:Nantradols_structure.png|300px|thumb|left|Desacetyllevonantradol, 80286-75-5<ref>{{pubchem|5488671}}</ref> (top left), ''N''-methyl-DALN (top right), and tetracyclic derivative (bottom)]]{{clear left}} |
|||
| ⚫ | |||
| ⚫ | Levonantradol has been clinically tested in cancer patients for its pain relief and antiemetic benefits. Cancer patients that endure chemotherapy often develop intense nausea, and Levonantradol has been tested to reduce these emetic symptoms. It is often used instead of THC because it has a higher efficacy. Levonantradol also acts on pain pathways in the central nervous system, which enables the drug to alleviate pain. Studies have shown an absence of emetic side effects within the half-life of the Levonantradol administered. Other studies suggest that cannabinoid agonists can synergize opioid anti-nociception. Cannabinoid receptors are located in nociceptive pathways, and CBs can promote signal transduction in TRP channels. Although Levonantradol relieves nociceptive and postoperative pain, decreases nausea, and improves spasticity in addition to being more effective than placebos, it has yet to be approved as legal medicine. Researchers have concluded that Levonantradol is no more effective than [[Codeine]], which is why they do not recommend expansion into clinical practice. |
||
* [[A-41988]] |
|||
* [[AM-919]] |
|||
* [[Nonabine]] |
|||
* [[PSB-SB-1202]] |
|||
==Notes== |
|||
'''Side Effects:''' |
|||
{{reflist}} |
|||
| ⚫ | The side effects for Levonantradol include ptosis, sedation, and ataxia in non-human primates. In rodents, the symptoms include dysphoria, memory impairment, motor incoordination, reduced concentration, and disorientation. Levonantradol also decreases startle response. In humans, side effects include dry mouth, drowsiness, dizziness, altered perception, mild sedation, and lack of concentration. It can cause an increase in heart rate and decrease in blood pressure. Euphoric symptoms rarely occurred in subjects. |
||
| ⚫ | |||
* [[CP 47,497]] |
|||
== References == |
== References == |
||
{{ |
{{refbegin}} |
||
| ⚫ | |||
{{cite journal| |
* {{cite journal | vauthors = Hosking RD, Zajicek JP | title = Therapeutic potential of cannabis in pain medicine | journal = British Journal of Anaesthesia | volume = 101 | issue = 1 | pages = 59–68 | date = July 2008 | pmid = 18515270 | doi = 10.1093/bja/aen119 | doi-access = free }} |
||
* {{cite journal | vauthors = McCarthy LE, Borison HL | title = Antiemetic activity of N-methyllevonantradol and nabilone in cisplatin-treated cats | journal = Journal of Clinical Pharmacology | volume = 21 | issue = S1 | pages = 30S–37S | date = Aug–Sep 1981 | pmid = 6271834 | doi = 10.1002/j.1552-4604.1981.tb02570.x | s2cid = 37795897 }} |
|||
{{cite journal| |
* {{cite journal | vauthors = Milewich L, Gant NF, Schwarz BE, Chen GT, MacDonald PC | title = 5 alpha-Reductase activity in human placenta | journal = American Journal of Obstetrics and Gynecology | volume = 133 | issue = 6 | pages = 611–617 | date = March 1979 | pmid = 34324 | doi = 10.1016/0002-9378(79)90006-1 }} |
||
{{refend}} |
|||
| ⚫ | |||
{{cite journal|last=Milewich|first=L|coauthors=Gant, NF; Schwarz, BE; Chen, GT; MacDonald, PC|title=5 alpha-Reductase activity in human placenta.|journal=American journal of obstetrics and gynecology|date=1979 Mar 15|volume=133|issue=6|pages=611–7|pmid=34324}} |
|||
{{Cannabinoids}} |
{{Cannabinoids}} |
||
{{ |
{{Hallucinogens}} |
||
{{Cannabinoidergics}} |
|||
{{cannabinoid-stub}} |
|||
| ⚫ | |||
[[Category:Cannabinoids]] |
[[Category:Cannabinoids]] |
||
[[Category:Drugs developed by Pfizer]] |
|||
[[Category:Phenanthridines]] |
[[Category:Phenanthridines]] |
||
[[Category:Phenol ethers]] |
[[Category:Phenol ethers]] |
||
[[Category: |
[[Category:CB1 receptor agonists]] |
||
| ⚫ | |||
Latest revision as of 01:35, 16 August 2024
 | |
| Clinical data | |
|---|---|
| ATC code |
|
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C27H35NO4 |
| Molar mass | 437.580 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| (verify) | |
Levonantradol (CP 50,556-1) is a synthetic cannabinoid analog of dronabinol (Marinol) developed by Pfizer in the 1980s. It is around 30 times more potent than THC, and exhibits antiemetic and analgesic effects via activation of CB1 and CB2 cannabinoid receptors.[1] Levonantradol is not currently used in medicine as dronabinol or nabilone are felt to be more useful for most conditions, however it is widely used in research into the potential therapeutic applications of cannabinoids.[2][3][4]
Pharmacodynamics
[edit]Levonantradol is a full CB1 receptor agonist. Cannabinoid receptors belong to the superfamily of G-protein coupled receptors (GPCRs), and endogenous cannabinoids naturally activate GPCRs. GPCRs modulate the inhibition of adenylyl cyclase and accumulation of the second messenger, cyclic adenosine monophosphate (cAMP). The CB1 receptor is the most common GPCR in the central nervous system. The activation of CB1Rs decrease calcium conductance and increase potassium conductance in the brain. CB signaling naturally modulates synaptic transmission and mediates psychoactivity, and synthetic cannabinoids mimic these same actions. Although the efficacy of Levonantradol is dependent on the level of GCPR activity, Full agonists like Levonantradol have the ability to activate GPCRs and convert Gα into a high affinity state for GTP or low affinity state for GDP. Previous studies suggest that Levonantradol has a higher binding affinity and efficacy than other similar synthetic cannabinoids (e.g. Δ9-THC).
Pharmacokinetics
[edit]Although Levonantradol has been extensively tested on animals including cats, rodents, and non-human primates. It has also been tested among cancer patient populations in clinical trials. Levonantradol is most commonly administered intramuscularly (I.M.), however it can also be administered orally. The dosage can range from 0.25 mg-3.0 mg every 2–4 hours, and the half-life is 1–2 hours. In order to administer Levonantradol intramuscularly, the drug must be dissolved in 5% ethanol, 5% emulphur, and 90% sterile saline. Synthetic cannabinoids like Levonantradol readily cross the blood–brain barrier because they are highly lipophilic and have low molecular weights. Levonantradol's bioavailability is variable due to the first pass metabolism.
Treatment
[edit]Levonantradol has been clinically tested in cancer patients for its pain relief and antiemetic benefits. Cancer patients that endure chemotherapy often develop intense nausea, and Levonantradol has been tested to reduce these emetic symptoms. It is often used instead of THC because it has a higher efficacy. Levonantradol also acts on pain pathways in the central nervous system, which enables the drug to alleviate pain. Studies have shown an absence of emetic side effects within the half-life of the Levonantradol administered. Other studies suggest that cannabinoid agonists can synergize opioid anti-nociception. Cannabinoid receptors are located in nociceptive pathways, and CBs can promote signal transduction in TRP channels. Although Levonantradol relieves nociceptive and postoperative pain, decreases nausea, and improves spasticity in addition to being more effective than placebos, it has yet to be approved as legal medicine. Researchers have concluded that Levonantradol is no more effective than Codeine, which is why they do not recommend expansion into clinical practice.
Side effects
[edit]The side effects for Levonantradol include ptosis, sedation, and ataxia in non-human primates. In rodents, the symptoms include dysphoria, memory impairment, motor incoordination, reduced concentration, and disorientation. Levonantradol also decreases startle response. In humans, side effects include dry mouth, drowsiness, dizziness, altered perception, mild sedation, and lack of concentration. It can cause an increase in heart rate and decrease in blood pressure. Euphoric symptoms rarely occurred in subjects.
Synthesis
[edit]
Dane salt formation between 3,5-dimethoxyaniline and ethyl acetoacetate followed by borohydrate reduction gives synthon 1. The amino group is protected by rxn with ethyl chloroformate, the ester group is saponified, and then cyclodehydration with polyphosphoric acid leads to the dihydroquinoline ring system (2). Deblocking with HBr is followed by etherification of the nonchelated phenolic hydroxyl gives 3. Treatment with NaH and ethyl formate results in both N-formylation and C-formylation of the active methylene to give 4. Michael addition of methyl vinyl ketone (MVP) followed by successive base treatments to remove the activating C-formyl group and then to complete the Robinson annulation to give 5. Lithium in liquid ammonia reduces the olefinic linkage and successive acetylation and sodium borohydrate reductions complete the synthesis of nantradol (6).
Related compounds
[edit]Numerous other compounds similar to levonantradol were also developed at the same time, including CP 42,096, CP 47,497, CP 55,940 and CP 55,244. The desacetyl derivative of levonantradol (DALN or CP 54,939) and its N-methyl derivative, as well as the tetracyclic analogue all have similar activity to levonantradol itself.[7]
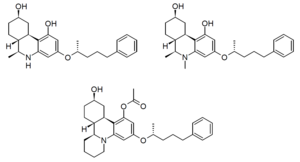
Desacetyllevonantradol, 80286-75-5[8] (top left), N-methyl-DALN (top right), and tetracyclic derivative (bottom)

See also
[edit]Notes
[edit]- ^ Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR (December 1988). "Pharmacology and stereoselectivity of structurally novel cannabinoids in mice". The Journal of Pharmacology and Experimental Therapeutics. 247 (3): 1046–1051. PMID 2849657.
- ^ Tramèr MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ (July 2001). "Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review". BMJ. 323 (7303): 16–21. doi:10.1136/bmj.323.7303.16. PMC 34325. PMID 11440936.
- ^ Campbell FA, Tramèr MR, Carroll D, Reynolds DJ, Moore RA, McQuay HJ (July 2001). "Are cannabinoids an effective and safe treatment option in the management of pain? A qualitative systematic review". BMJ. 323 (7303): 13–16. doi:10.1136/bmj.323.7303.13. PMC 34324. PMID 11440935.
- ^ Ben Amar M (April 2006). "Cannabinoids in medicine: A review of their therapeutic potential". Journal of Ethnopharmacology. 105 (1–2): 1–25. CiteSeerX 10.1.1.180.308. doi:10.1016/j.jep.2006.02.001. PMID 16540272.
- ^ Johnson MR, Milne GM (1980). "Recent discoveries in the search for non-opiate analgetics". Journal of Heterocyclic Chemistry. 17 (8): 1817–1820. doi:10.1002/jhet.5570170841.
- ^ Sheshenev AE, Boltukhina EV, Hii KK (May 2013). "Levonantradol: asymmetric synthesis and structural analysis". Chemical Communications. 49 (35): 3685–3687. doi:10.1039/C3CC41388H. PMID 23535893.
- ^ Howlett AC, Johnson MR, Melvin LS, Milne GM (March 1988). "Nonclassical cannabinoid analgetics inhibit adenylate cyclase: development of a cannabinoid receptor model". Molecular Pharmacology. 33 (3): 297–302. PMID 3352594.
- ^ CID 5488671 from PubChem
References
[edit]- Childers SR (March 2006). "Activation of G-proteins in brain by endogenous and exogenous cannabinoids". The AAPS Journal. 8 (1): E112–E117. doi:10.1208/aapsj080113. PMC 2751429. PMID 16584117.
- Hosking RD, Zajicek JP (July 2008). "Therapeutic potential of cannabis in pain medicine". British Journal of Anaesthesia. 101 (1): 59–68. doi:10.1093/bja/aen119. PMID 18515270.
- McCarthy LE, Borison HL (Aug–Sep 1981). "Antiemetic activity of N-methyllevonantradol and nabilone in cisplatin-treated cats". Journal of Clinical Pharmacology. 21 (S1): 30S–37S. doi:10.1002/j.1552-4604.1981.tb02570.x. PMID 6271834. S2CID 37795897.
- Milewich L, Gant NF, Schwarz BE, Chen GT, MacDonald PC (March 1979). "5 alpha-Reductase activity in human placenta". American Journal of Obstetrics and Gynecology. 133 (6): 611–617. doi:10.1016/0002-9378(79)90006-1. PMID 34324.