
Aryne: Difference between revisions
mNo edit summary |
|||
| Line 40: | Line 40: | ||
In 2015, scientists from IBM Research and from [[CiQUS]] at the [[University of Santiago de Compostela]], Spain, have confirmed the existence and characterized the structure of arynes by publishing the imaging of a single aryne molecule for the first time using [[Scanning tunneling microscope|STM]].<ref>On-surface generation and imaging of arynes by atomic force microscopy, D.Pérez, E.Guitián, D.Peña, L.Gross, ''Nature Chemistry'', '''2015''', 7, 623</ref> |
In 2015, scientists from IBM Research and from [[CiQUS]] at the [[University of Santiago de Compostela]], Spain, have confirmed the existence and characterized the structure of arynes by publishing the imaging of a single aryne molecule for the first time using [[Scanning tunneling microscope|STM]].<ref>On-surface generation and imaging of arynes by atomic force microscopy, D.Pérez, E.Guitián, D.Peña, L.Gross, ''Nature Chemistry'', '''2015''', 7, 623</ref> |
||
==Generation of arynes<ref>{{Cite book|title=Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds (J. Mortier Ed.)|last=The Chemistry of Arynes: An Overview|first=Chapter 12|last2=Sanz|first2=R.|last3=Suarez|first3=A.|publisher=John Wiley & Sons|year=2016|isbn=978-1-118-75498-6|location= |
==Generation of arynes<ref>{{Cite book|title=Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds (J. Mortier Ed.)|last=The Chemistry of Arynes: An Overview|first=Chapter 12|last2=Sanz|first2=R.|last3=Suarez|first3=A.|publisher=John Wiley & Sons|year=2016|isbn=978-1-118-75498-6|location=|pages=301-336|doi=10.1002/9781118754887}}</ref>== |
||
Due to extreme reactivity of arynes, they must be generated ''in situ''. The [[reactive intermediate]] rapidly [[dimer (chemistry)|dimerises]] to [[biphenylene]]. |
Due to extreme reactivity of arynes, they must be generated ''in situ''. The [[reactive intermediate]] rapidly [[dimer (chemistry)|dimerises]] to [[biphenylene]]. |
||
Revision as of 14:54, 7 September 2018
Arynes[1] or benzynes[2] are highly reactive species derived from an aromatic ring by removal of two ortho substituents.[3][4] Arynes are usually best described as having a strained triple bond; however, they possess some biradical character as well. They are thus 1,2-didehydroarenes. However, the terms aryne and benzyne are sometimes used to refer to isomers of dehydroarene as well.[5] as well.[6]
The discovery of benzyne led to rapid developments in synthetic methodologies to make this highly reactive intermediate useful for organic synthesis. A variety of natural products have been prepared using arynes as intermediates.[7]
Bonding
The alkyne representation of benzyne is the most widely encountered, however, the cumulene and diradical structures are significant resonance contributors.[8]

Geometric constraints on the triple bond in ortho-benzyne result in reduced overlap of in-plane p-orbitals, and thus weaker triple bond.[9] The vibrational frequency of the triple bond in benzyne was assigned by Rasziszhewski to be 1846 cm −1,[10] indicating a much weaker triple bond than in unstrained alkyne with vibrational frequency of approximately 2150 cm−1. Nevertheless, ortho-benzyne is more like a strained alkyne than a biradical, as seen from the large singlet–triplet gap and alkyne-like reactivity.[11] Geometric restrictions also result in significant lowering of the energy the LUMO of arynes (6.41 eV in 2-butyne to 1.33 eV in benzyne) while the energy of the HOMO remains essentially unchanged according to computations.[9][12]

LUMO orbital of aryne lies much lower than LUMO of unstrained alkynes, which makes it a better energy match for HOMO of nucleophiles. Hence, benzyne possesses electrophilic character and undergoes reactions with nucleophiles.[13] A detailed MO analysis of benzyne was presented in 1968.[14]

History
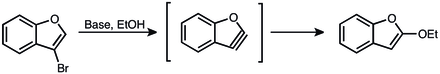
The first hint of the existence of aryne intermediate came from the work of Stoermer and Kahlert in 1902. They observed that upon treatment of 3-bromobenzofuran with base in ethanol 2-ethoxybenzofuran is formed. Based on this observation they postulated an aryne intermediate.[15]
Georg Wittig and coworkers proposed that the formation of biphenyl via reactions of fluorobenzene and phenyllithium proceeded via a zwitterionic intermediate.[16][17][18] and experimentally confirmed[clarification needed] by John D. Roberts in 1953.[19][20][21][22][23]

In 1953 John D. Roberts performed the classic 14C labeling experiment, which provided strong support for benzyne.[19] Roberts and his students performed the reaction of chlorobenzene-1-14C with potassium amide, and analyzed the 14C-label incorporation into the resulting aniline: equal amounts of aniline with 14C incorporation at C-1 and C-2 were observed. This result necessitated a symmetrical intermediate – now known as benzyne.

Soon after Roberts’ discovery, Wittig and Pohmer found that benzyne can participate in [4+2] cycloaddition reactions.[24]

Additional evidence for the existence of benzyne came from spectroscopic studies: IR,[25] UV/Vis,[26] microwave,[27] and NMR spectroscopies.[28][29]
Benzyne has been observed in a "molecular container".[29][30]
In 2015, scientists from IBM Research and from CiQUS at the University of Santiago de Compostela, Spain, have confirmed the existence and characterized the structure of arynes by publishing the imaging of a single aryne molecule for the first time using STM.[31]
Generation of arynes[32]
Due to extreme reactivity of arynes, they must be generated in situ. The reactive intermediate rapidly dimerises to biphenylene.
In the early days of benzyne chemistry, harsh conditions were needed to generate arynes – many of the methods require strong base or high temperatures. Aryl halide can be treated with strong base to remove an aromatic proton and generate benzyne via elimination.

Arenediazonium-2-carboxylates can serve as precursors to benzynes. The main drawback of this method is the explosive nature of diazonium compounds. As an example, anthranilic acid can be converted to the zwitterionic benzene-2-carboxylate which, on heating, forms benzyne.[33]

Milder methods for benzyne generation have been developed. Aryl triflates have been widely used in synthesis.[7] Fluoride displacement of the trimethylsilyl group, as shown below, allows for generation of benzyne under mild conditions.

A hexadehydro Diels-Alder reaction (HDDA) involves cycloaddition of 1,3-diyne and alkyne.[34]

N-amination of 1H-benzotriazole with hydroxylamine-O-sulfonic acid generates an intermediate which can be oxidised to benzyne in almost quantitative yield with lead(IV) acetate.[35]

Reactions of arynes
Even at low temperatures arynes are extremely reactive. Their reactivity can be classified in four major classes: (1) nucleophilic additions, (2) pericyclic reactions, (3) bond-insertion, and (4) metal-catalyzed reactions.
Nucleophilic additions to arynes
Upon treatment with basic nucleophiles, aryl halides deprotonate alpha to the leaving group, resulting in dehydrohalogenation. The resulting benzyne forms addition products, usually by initial protonation. Generation of the benzyne intermediate is the slow step in the reaction.[36]

"Aryne coupling" reactions allow for generation of biphenyl compounds which are valuable in pharmaceutical industry, agriculture and as ligands in many metal-catalyzed transformations.[37]

The metal–arene product can also add to another aryne, leading to chain-growth polymerization. Using copper(I) cyanide as the initiator to add to the first aryne yielded polymers containing up to about 100 arene units.[38]
Substituent effect
Regiochemistry of the triple bond formation
When leaving group (LG) and substituent (Y) are mutually ortho or para, only one benzyne intermediate is possible. However, when LG is meta to Y, then regiochemical outcomes (A and B) are possible. If Y is electron withdrawing, then HB is more acidic than HA resulting in regioisomer B being generated. Analogously, if Y is electron donating, regioisomer A is generated, since now HA is the more acidic proton.

Regiochemistry of the addition of nucleophile to the triple bond
There are two possible regioisomers of benzyne with substituent (Y): triple bond can be positioned between C2 and C3 or between C3 and C4. Substituents ortho to the leaving group will lead to the triple bond between C2 and C3. Para Y and LG will lead to regioisomer with triple bond between C3 and C4. Meta substituent can afford both regioisomers as described above. In case of triple bond located between C2 and C3, electron withdrawing substituents, e.g. CF3, (EWG) will direct the nucleophile addition to place carbanion as close as possible to EWG. However, electron donating substituents, e.g. CH3, (EDG) will provide little selectivity between products. In the regioisomer where triple bond is located between C3 and C4 the effect of substituent on nucleophile addition is diminished, and mixtures of para and meta products are often obtained.[36]

Examples in total synthesis
Nucleophilic additions to arynes have been widely used in natural product total synthesis. Indeed, nucleophilic additions of arynes are some of the oldest known applications of aryne chemistry.[7] Nucleophilic addition to aryne was used in the attempted synthesis of cryptaustoline (1) and cryptowoline (2).[39]

The synthesis of the tetracyclic meroterpenoid (+)-liphagal involved an aryne intermediate.[40] Their approach employed an aryne cyclization to close the final ring of the natural product.[7]

Multicomponent reactions of arynes are powerful transformations that allow for rapid formation of 1,2-disubsituted arenes. Despite their potential utility, examples of multicomponent aryne reactions in natural product synthesis are scarce.[7] A four-component aryne coupling reaction was employed in the synthesis of dehydroaltenuene B.[41]

Pericyclic reactions of arynes
Dimerization and trimerization
Benzyne undergoes rapid dimerization to form biphenylene. Some routes to benzyne lead to especially rapid and high yield of this subsequent reaction.[33][35] Trimerization gives triphenylene.[42]
[4+2] cycloadditions
Benzynes can undergo [4+2] cyclization reactions. The concerted mechanism of the Diels-Alder reaction between benzyne and furan is shown below. However, many benzyne [4+2] cycloadditions are thought to proceed via a stepwise mechanism.
A classic example is the synthesis of 1,2,3,4-tetraphenylnaphthalene.[43] Tetrabromobenzene can react with butyllithium and furan to form a tetrahydroanthracene [44][45]

[4+2] cycloadditions of arynes have been commonly applied to natural product total synthesis. The main limitation of such approach, however, is the need to use constrained dienes, such as furan and cyclopentadiene.[7] In 2009 Buszek and co-workers synthesized Herbindole A using aryne [4+2]-cycloaddition.[46] 6,7-indolyne undergoes [4+2] cycloaddition with cyclopentadiene to afford complex tetracyclic product.

[2+2] cycloadditions
Benzynes undergo [2+2] cycloaddition with a wide range of olefins. Due to electrophilic nature of benzyne, olefins bearing electron-donating substituents work best for this reaction.[47]
Due to significant byproduct formation, aryne [2+2] chemistry is rarely utilized in natural product total synthesis.[7] Nevertheless, several examples do exist. In 1982, Stevens and co-workers reported a synthesis of taxodione that utilized [2+2] cycloaddition between an aryne and a ketene acetal.[48]

Bond-insertion reactions of arynes
The first example of aryne σ-bond insertion reaction is the synthesis of Melleine in 1973.[49]

Metal-catalyzed reactions of arynes
Mori and co-workers performed a palladium-catalyzed [2+2+2]-cocyclization of aryne and diyne in their total synthesis of taiwanins C.[50]

Other dehydrobenzenes
If benzyne is 1,2-didehydrobenzene, two further isomers are possible: 1,3-didehydrobenzene and 1,4-didehydrobenzene.[11] Their energies in silico are, respectively, 106, 122, and 138 kcal/mol (444, 510, and 577 kJ/mol).[51]

The interconversion of the 1,2-, 1,3- and 1,4-didehydrobenzenes has been studied.[51][52] A 1,2- to 1,3-didehydrobenzene conversion has been postulated to occur in the pyrolysis (900 °C) of the phenyl substituted aryne precursors [51] as shown below. Extremely high temperatures are required for benzyne interconversion.

1,4-Didehydroarenes
In classical 1,4-didehydrobenzene experiments, heating to 300 °C, [1,6-D2]-A readily equilibrates with [3,2-D2]-B, but does not equilibrate with C or D. The simultaneous migration of deuterium atoms to form B, and the fact that none of C or D is formed can only be explained by a presence of a cyclic and symmetrical intermediate –1,4-didehydrobenzene.[53]

The Bergman cyclization proved to be even more important with the discovery of enediyne "cytostatics", compounds able to cleave double-stranded DNA. One example of enediyne cytostatic is calicheamicin, which in cell reveals a 1,4-didehydrobenzene moiety. The ability of substituted 1,4-didehydrobenzene to abstract hydrogen atoms from DNA is thought to be a key step in the biological activity of the potential antitumor/antibiotic enediyne drugs, such as calicheamicin. However, enediyne molecules tend to be very reactive and nonselective, making them poor drug candidates. Computational chemistry helped predict the behavior of 1,4-didehydrobenzene analogues that in future might help produce a drug candidates with ameliorated reactivity and high selectivity.[54]
Two states were proposed for 1,4-didehydrobenzene: singlet and triplet, with the singlet state lower in energy.[55][56] Triplet state represents two noninteracting radical centers, and hence should abstract hydrogens at the same rate as phenyl radical. However, singlet state is more stabilized than the triplet, and therefore some of the stabilizing energy will be lost in order to form the transition state for hydrogen cleavage, leading to slower hydrogen abstraction. Chen proposed the use of 1,4-didehydrobenzene analogues that have large singlet-triplet energy gaps to enhance selectivity of enediyne drug candidates.[57]
See also
- More examples use of aryne chemistry: tricyclobutabenzene, in-methylcyclophane, Transition metal benzyne complex
- The pyridine equivalent pyridyne
References
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Aryne". doi:10.1351/goldbook.A00465
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Benzynes". doi:10.1351/goldbook.B00634
- ^ Gilchrist T.C.; Rees C.W.; (1969) Carbenes, Nitrenes and Arynes Nelson. London.
- ^ The Benzyne and Related Intermediates. H. Heaney Chem. Rev., 1962, 62 (2), pp 81–97 doi:10.1021/cr60216a001
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Dehydroarenes". doi:10.1351/goldbook.D01574
- ^ IUPAC Gold Book entry for "benzynes" identifies "m-benzyne" and "p-benzyne" as erroneous terms for 1,3- and 1,4-didehydrobenzene
- ^ a b c d e f g Tadross, P. M.; Stoltz, B. M. A Comprehensive History of Arynes in Natural Product Total SynthesisChem. Rev. 2012, 112, 3550
- ^ Anslyn, E. V.; Dougherty, D. A.: Modern Physical Organic Chemistry, University Science Books, 2006, p612.
- ^ a b Gampe, C. M.; Carreira, E. M. Angew Chem. Int. Ed. 2012, 51, 3766
- ^ Rasziszewski, J. G.; Hess. B. A. J.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52
- ^ a b Wenk, H. H.; Winkler, M.; Sander, W. Angew. Chem. Int. Ed. 2003, 42, 502
- ^ Rondan, N. G.; Domelsmith, L. N.; Houk, K.N. Tetrahedron Lett. 1979, 20, 3273
- ^ Gilchrist, T. L. Supplement C: The Chemistry of Triple Bonded Functional Groups, Part 1. Patai, S.; Rappaport, Z. Eds., John Wiley & Sons, New York, 1983
- ^ Hoffmann, R.; Imamura, A.;Hehre, W. J. J. Am. Chem. Soc. 1968, 90, 1499
- ^ "Ueber das 1- und 2-Brom-cumaron". doi:10.1002/cber.19020350286. Retrieved 27 June 2013.
{{cite journal}}: Cite journal requires|journal=(help); Unknown parameter|deadurl=ignored (|url-status=suggested) (help) - ^ Wittig, G., Pieper, G. and Fuhrmann, G. (1940), Über die Bildung von Diphenyl aus Fluorbenzol und Phenyl-lithium (IV. Mitteil. über Austauschreaktionen mit Phenyl-lithium). Berichte der deutschen chemischen Gesellschaft (A and B Series), 73: 1193–1197. doi:10.1002/cber.19400731113
- ^ Phenyl-lithium, der Schlüssel zu einer neuen Chemie metallorganischer Verbindungen Georg Wittig Naturwissenschaften, 1942, Volume 30, Numbers 46-47, Pages 696-703 doi:10.1007/BF01489519
- ^ Wittig, G. (1954), Fortschritte auf dem Gebiet der organischen Aniono-Chemie. Angewandte Chemie, 66: 10–17. doi:10.1002/ange.19540660103
- ^ a b rearrangement in the reaction of chlorobenzene-1-C14 with potassium amid John D. Roberts, Howard E. Simmons Jr., L. A. Carlsmith, C. Wheaton Vaughan J. Am. Chem. Soc., 1953, 75 (13), pp 3290–3291 doi:10.1021/ja01109a523
- ^ The Mechanism of Aminations of Halobenzenes John D. Roberts, Dorothy A. Semenow, Howard E. Simmons Jr., L. A. Carlsmith J. Am. Chem. Soc., 1956, 78 (3), pp 601–611 doi:10.1021/ja01584a024
- ^ Orientation in Aminations of Substituted Halobenzenes John D. Roberts, C. Wheaton Vaughan, L. A. Carlsmith, Dorothy A. Semenow J. Am. Chem. Soc., 1956, 78 (3), pp 611–614 doi:10.1021/ja01584a025
- ^ Modern Arylation Methods. Edited by Lutz Ackermann 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN 978-3-527-31937-4
- ^ The Benzyne and Related Intermediates. H. Heaney Chem. Rev., 1962, 62 (2), pp 81–97 doi:10.1021/cr60216a001
- ^ Wittig, G.; Pohmer, L. Angew. Chem. 1955, 67(13), 348.
- ^ Radziszewski, J. G.; Hess, Jr. B. A.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52.
- ^ Wenthold, P. G.; Squires, R. R.; Lineberger, W. C. J. Am. Chem. Soc. 1998, 120, 5279
- ^ Kukolich, S. G.; Tanjaroon,C.; McCarthy, M. C.; Thaddeus, P. J. Chem. Phys. 2003, 119, 4353
- ^ Orendt, A. M.; Facelli, J. C.; Radziszewski, J. G.; Horton, W. J.; Grant, D. M.; Michl, J. J. Am. Chem. Soc. 1996, 118, 846
- ^ a b Warmuth,R. Angew. Chem. Int. Ed. Engl. 1997, 36, 1347
- ^ Warmuth, R.; Yoon, Acc. Chem. Res. 2001, 34, 96
- ^ On-surface generation and imaging of arynes by atomic force microscopy, D.Pérez, E.Guitián, D.Peña, L.Gross, Nature Chemistry, 2015, 7, 623
- ^ The Chemistry of Arynes: An Overview, Chapter 12; Sanz, R.; Suarez, A. (2016). Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds (J. Mortier Ed.). John Wiley & Sons. pp. 301–336. doi:10.1002/9781118754887. ISBN 978-1-118-75498-6.
{{cite book}}: CS1 maint: numeric names: authors list (link) - ^ a b Logullo, Francis M.; Seitz, Arnold M.; Friedman, Lester (1968). "Benzenediazonium-2-Carboxylate and Biphenylene (Benzenediazonium, o-carboxy-, hydroxide, inner salt)". Organic Syntheses. 48: 12. doi:10.15227/orgsyn.048.0012; Collected Volumes, vol. 5, p. 54.
- ^ Hoye, T. R.; Baire, B.; Niu, D.; Willoughby, P. H.; Woods, B. P. Nature, 2012, 490, 208
- ^ a b Campbell, C.D.; Rees, C.W. (1969). "Reactive intermediates. Part I. Synthesis and oxidation of 1- and 2-aminobenzotriazole". J. Chem. Soc. C. 1969 (5): 742–747. doi:10.1039/J39690000742.
- ^ a b Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry. University Science Books, 2006.
- ^ Diemer, V.; Begaut, M.; Leroux, F. R.; Colobert, F. Eur. J. Org. Chem. 2011, 341
- ^ Mizukoshi, Yoshihide; Mikami, Koichiro; Uchiyama, Masanobu (2015). "Aryne Polymerization Enabling Straightforward Synthesis of Elusive Poly(ortho-arylene)s". J. Am. Chem. Soc. 137 (1): 74–77. doi:10.1021/ja5112207.
- ^ Kametani, T.; Ogasawara, K. J. J. Chem. Soc., C 1967, 2208
- ^ Day, J. J.; McFadden, R. M.; Virgil, S. C.; Kolding, H.; Alleva, J. L.; Stoltz, B. M. Angew. Chem. Int. Ed. 2011, 50, 6814.
- ^ Soorukram, D.; Qu, T.; Barrett, A. G. M. Org. Lett. 2008, 10, 3833
- ^ Heaney, H.; Millar, I. T. (1960). "Triphenylene". Organic Syntheses. 40: 105; Collected Volumes, vol. 5, 1973, p. 1120.
- ^ Organic Syntheses, Coll. Vol. 5, p.1037 (1973); Vol. 46, p.107 (1966). Link
- ^ Organic Syntheses, Coll. Vol. 10, p.678; Vol. 75, p.201 Article
- ^ The mixture of syn and anti stereoisomers can be separated based on difference in methanol solubility.
- ^ Buszek, K. R.; Brown, N.; Kuo, D. Org. Lett. 2009, 11, 201
- ^ Pellissier, H.; Santelli, M. Tetrahedron, 2003, 59, 701
- ^ Stevens, R. V.; Bisacchi, G. S> J. Org, Chem. 1982, 47, 2396
- ^ Guyot, M.; Molho, D. Tetrahedron Lett. 1973, 14, 3433
- ^ Sato, Y.; Tamura,T.; Mori, M. Angew. Chem. Int. Ed. 2004, 43, 2436
- ^ a b c A m-Benzyne to o-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Blake, M. E.; Bartlett, K. L.; Jones, M. Jr. J. Am. Chem. Soc. 2003, 125, 6485. doi:10.1021/ja0213672
- ^ A p-Benzyne to m-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Completion of the Benzyne Cascade, Polishchuk, A. L.; Bartlett, K. L.; Friedman, L. A.; Jones, M. Jr. J. Phys. Org. Chem. 2004, Volume 17, Issue 9 , Pages 798 - 806. doi:10.1002/poc.797
- ^ Jones, R. R,; Bergman, R. G. J. Am. Chem. Soc. 1972, 94, 660
- ^ Bachrach, S. M. Computational Organic Chemistry. John Wiley & Sons, Inc. 2007
- ^ Clauberg, H.; Minsek, D. W.; Chen, P. J. Am. Chem. Soc. 1992, 114, 99.
- ^ Blush, J. A.; Clauberg, H.; Kohn, D. W.; Minsek, D. W.; Zhang, X.; Chen, P. Acc. Chem. Res. 1992, 25, 385
- ^ Chen, P. Angew. Chem. Int. Ed. Engl. 1996, 35, 1478.
| Authority control databases: National |
|---|