
ATAC-seq: Difference between revisions
Jrahimik1986 (talk | contribs) |
m v2.03b - Bot T20 CW#61 - WP:WCW project (Reference before punctuation) |
||
| Line 13: | Line 13: | ||
== Single-cell ATAC-seq == |
== Single-cell ATAC-seq == |
||
Modifications to the ATAC-seq protocol have been made to accommodate [[single-cell analysis]]. [[Microfluidics]] can be used to separate single nuclei and perform ATAC-seq reactions individually.<ref name="BuenrostroWuLitzenburger">{{cite journal | vauthors = Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ | display-authors = 6 | title = Single-cell chromatin accessibility reveals principles of regulatory variation | journal = Nature | volume = 523 | issue = 7561 | pages = 486–90 | date = July 2015 | pmid = 26083756 | pmc = 4685948 | doi = 10.1038/nature14590 | bibcode = 2015Natur.523..486B }}</ref> With this approach, single cells are captured by either a microfluidic device or a liquid deposition system before tagmentation.<ref name="BuenrostroWuLitzenburger" /><ref name="MezgerKlemm2018">{{cite journal | vauthors = Mezger A, Klemm S, Mann I, Brower K, Mir A, Bostick M, Farmer A, Fordyce P, Linnarsson S, Greenleaf W | display-authors = 6 | title = High-throughput chromatin accessibility profiling at single-cell resolution | journal = Nature Communications | volume = 9 | issue = 1 | pages = 3647 | date = September 2018 | pmid = 30194434 | pmc = 6128862 | doi = 10.1038/s41467-018-05887-x | bibcode = 2018NatCo...9.3647M }}</ref> An alternative technique that does not require single cell isolation is combinatorial cellular indexing<ref>{{cite journal |last1=Cusanovich |first1=Darren |title=Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing |journal=Science |date=May 2015 |volume=348 |issue=6237 |pages=910–914 |doi=10.1126/science.aab1601 |pmid=25953818 |pmc=4836442}}</ref> |
Modifications to the ATAC-seq protocol have been made to accommodate [[single-cell analysis]]. [[Microfluidics]] can be used to separate single nuclei and perform ATAC-seq reactions individually.<ref name="BuenrostroWuLitzenburger">{{cite journal | vauthors = Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ | display-authors = 6 | title = Single-cell chromatin accessibility reveals principles of regulatory variation | journal = Nature | volume = 523 | issue = 7561 | pages = 486–90 | date = July 2015 | pmid = 26083756 | pmc = 4685948 | doi = 10.1038/nature14590 | bibcode = 2015Natur.523..486B }}</ref> With this approach, single cells are captured by either a microfluidic device or a liquid deposition system before tagmentation.<ref name="BuenrostroWuLitzenburger" /><ref name="MezgerKlemm2018">{{cite journal | vauthors = Mezger A, Klemm S, Mann I, Brower K, Mir A, Bostick M, Farmer A, Fordyce P, Linnarsson S, Greenleaf W | display-authors = 6 | title = High-throughput chromatin accessibility profiling at single-cell resolution | journal = Nature Communications | volume = 9 | issue = 1 | pages = 3647 | date = September 2018 | pmid = 30194434 | pmc = 6128862 | doi = 10.1038/s41467-018-05887-x | bibcode = 2018NatCo...9.3647M }}</ref> An alternative technique that does not require single cell isolation is combinatorial cellular indexing.<ref>{{cite journal |last1=Cusanovich |first1=Darren |title=Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing |journal=Science |date=May 2015 |volume=348 |issue=6237 |pages=910–914 |doi=10.1126/science.aab1601 |pmid=25953818 |pmc=4836442}}</ref> This technique uses [[DNA barcoding|barcoding]] to measure chromatin accessibility in thousands of individual cells; it can generate epigenomic profiles from 10,000-100,000 cells per experiment.<ref name="LareauDuarte2019">{{cite journal | vauthors = Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, Pokholok D, Aryee MJ, Steemers FJ, Lebofsky R, Buenrostro JD | display-authors = 8 |year=2019 |title=Droplet-based combinatorial indexing for massive scale single-cell epigenomics |journal=bioRxiv |volume= |pages= |doi=10.1101/612713 |doi-access=free }}</ref> But combinatorial cellular indexing requires additional, custom-engineered equipment or a large quantity of custom, modified Tn5.<ref name="ChenMiragaia2018">{{cite journal | vauthors = Chen X, Miragaia RJ, Natarajan KN, Teichmann SA | title = A rapid and robust method for single cell chromatin accessibility profiling | journal = Nature Communications | volume = 9 | issue = 1 | pages = 5345 | date = December 2018 | pmid = 30559361 | pmc = 6297232 | doi = 10.1038/s41467-018-07771-0 | bibcode = 2018NatCo...9.5345C }}</ref> |
||
Computational analysis of scATAC-seq is based on construction of a count matrix with number of reads per open chromatin regions. Open chromatin regions can be defined, for example, by standard peak calling of pseudo bulk ATAC-seq data. Further steps include data reduction with PCA and clustering of cells<ref name="MezgerKlemm2018" /> |
Computational analysis of scATAC-seq is based on construction of a count matrix with number of reads per open chromatin regions. Open chromatin regions can be defined, for example, by standard peak calling of pseudo bulk ATAC-seq data. Further steps include data reduction with PCA and clustering of cells.<ref name="MezgerKlemm2018" /> scATAC-seq matrices can be extremely large (hundreds of thousands of regions) and is extremely sparse, i.e. less than 3% of entries are non-zero.<ref name=":0">{{cite journal |last1=Li |first1=Zhijian |last2=Kuppe |first2=Christoph |last3=Cheng |first3=Mingbo |last4=Menzel |first4=Sylvia |last5=Zenke |first5=Martin |last6=Kramann |first6=Rafael |last7=Costa |first7=Ivan G. |name-list-format = vanc | display-authors = 6 |date=2019-12-05|title=scOpen: chromatin-accessibility estimation of single-cell ATAC data|url=https://www.biorxiv.org/content/10.1101/865931v1|journal=bioRxiv|language=en|pages=865931|doi=10.1101/865931|doi-access=free }}</ref> Therefore, imputation of count matrix is another crucial step. As with bulk ATAC-seq, scATAC-seq allows finding regulators like transcription factors controlling gene expression of cells. This can be achieved by looking at the number of reads around TF motifs<ref>{{cite journal | vauthors = Schep AN, Wu B, Buenrostro JD, Greenleaf WJ | title = chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data | journal = Nature Methods | volume = 14 | issue = 10 | pages = 975–978 | date = October 2017 | pmid = 28825706 | pmc = 5623146 | doi = 10.1038/nmeth.4401 }}</ref> or footprinting analysis.<ref name=":0" /> |
||
== References == |
== References == |
||
Revision as of 08:15, 25 September 2020
ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is a technique used in molecular biology to assess genome-wide chromatin accessibility.[1] In 2013, the technique was first described as an alternative advanced method for MNase-seq, FAIRE-Seq and DNase-Seq.[1] ATAC-seq is a faster and more sensitive analysis of the epigenome than DNase-seq or MNase-seq.[2][3][4]
Description
ATAC-seq identifies accessible DNA regions by probing open chromatin with hyperactive mutant Tn5 Transposase that inserts sequencing adapters into open regions of the genome. [2][5] While naturally occurring transposases have a low level of activity, ATAC-seq employs the mutated hyperactive transposase.[6] In a process called "tagmentation", Tn5 transposase cleaves and tags double-stranded DNA with sequencing adaptors.[7][8] The tagged DNA fragments are then purified, PCR-amplified, and sequenced using next-generation sequencing.[8] Sequencing reads can then be used to infer regions of increased accessibility as well as to map regions of transcription factor binding sites and nucleosome positions.[2] The number of reads for a region correlate with how open that chromatin is, at single nucleotide resolution.[2] ATAC-seq requires no sonication or phenol-chloroform extraction like FAIRE-seq;[9] no antibodies like ChIP-seq;[10] and no sensitive enzymatic digestion like MNase-seq or DNase-seq.[11] ATAC-seq preparation can be completed in under three hours.[12]
Applications
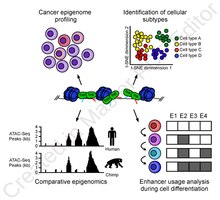
ATAC-Seq analysis is used to investigate a number of chromatin-accessibility signatures. The most common use is nucleosome mapping experiments,[3] but it can be applied to mapping transcription factor binding sites,[13] adapted to map DNA methylation sites,[14] or combined with sequencing techniques.[15]
The utility of high-resolution enhancer mapping ranges from studying the evolutionary divergence of enhancer usage (e.g. between chimps and humans) during development[16] and uncovering a lineage-specific enhancer map used during blood cell differentiation.[17]
ATAC-Seq has also been applied to defining the genome-wide chromatin accessibility landscape in human cancers,[18] and revealing an overall decrease in chromatin accessibility in macular degeneration.[19] Computational footprinting methods can be performed on ATAC-seq to find cell specific binding sites and transcription factors with cell specific activity.[13]
Single-cell ATAC-seq
Modifications to the ATAC-seq protocol have been made to accommodate single-cell analysis. Microfluidics can be used to separate single nuclei and perform ATAC-seq reactions individually.[12] With this approach, single cells are captured by either a microfluidic device or a liquid deposition system before tagmentation.[12][20] An alternative technique that does not require single cell isolation is combinatorial cellular indexing.[21] This technique uses barcoding to measure chromatin accessibility in thousands of individual cells; it can generate epigenomic profiles from 10,000-100,000 cells per experiment.[22] But combinatorial cellular indexing requires additional, custom-engineered equipment or a large quantity of custom, modified Tn5.[23]
Computational analysis of scATAC-seq is based on construction of a count matrix with number of reads per open chromatin regions. Open chromatin regions can be defined, for example, by standard peak calling of pseudo bulk ATAC-seq data. Further steps include data reduction with PCA and clustering of cells.[20] scATAC-seq matrices can be extremely large (hundreds of thousands of regions) and is extremely sparse, i.e. less than 3% of entries are non-zero.[24] Therefore, imputation of count matrix is another crucial step. As with bulk ATAC-seq, scATAC-seq allows finding regulators like transcription factors controlling gene expression of cells. This can be achieved by looking at the number of reads around TF motifs[25] or footprinting analysis.[24]
References
- ^ a b Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (December 2013). "Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position". Nature Methods. 10 (12): 1213–8. doi:10.1038/nmeth.2688. PMC 3959825. PMID 24097267.
- ^ a b c d Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (January 2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109: 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. PMC 4374986. PMID 25559105.
- ^ a b Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ (November 2015). "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research. 25 (11): 1757–70. doi:10.1101/gr.192294.115. PMC 4617971. PMID 26314830.
- ^ Song L, Crawford GE (February 2010). "DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. PMC 3627383. PMID 20150147.
- ^ Bajic, Marko; Maher, Kelsey A.; Deal, Roger B. (2018). "Identification of Open Chromatin Regions in Plant Genomes Using ATAC-Seq". Plant Chromatin Dynamics. Methods in Molecular Biology. Vol. 1675. pp. 183–201. doi:10.1007/978-1-4939-7318-7_12. ISBN 978-1-4939-7317-0. ISSN 1064-3745. PMC 5693289. PMID 29052193.
{{cite book}}: Unknown parameter|name-list-format=ignored (|name-list-style=suggested) (help) - ^ Reznikoff WS (2008). "Transposon Tn5". Annual Review of Genetics. 42 (1): 269–86. doi:10.1146/annurev.genet.42.110807.091656. PMID 18680433.
- ^ Adey, Andrew (December 2010). "Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition". Genome Biology. 11 (12): R119. doi:10.1186/gb-2010-11-12-r119. PMC 3046479. PMID 21143862.
{{cite journal}}: CS1 maint: unflagged free DOI (link) - ^ a b Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, Sandberg R (December 2014). "Tn5 transposase and tagmentation procedures for massively scaled sequencing projects". Genome Research. 24 (12): 2033–40. doi:10.1101/gr.177881.114. PMC 4248319. PMID 25079858.
- ^ Simon JM, Giresi PG, Davis IJ, Lieb JD (January 2012). "Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA". Nature Protocols. 7 (2): 256–67. doi:10.1038/nprot.2011.444. PMC 3784247. PMID 22262007.
- ^ Savic D, Partridge EC, Newberry KM, Smith SB, Meadows SK, Roberts BS, et al. (October 2015). "CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins". Genome Research. 25 (10): 1581–9. doi:10.1101/gr.193540.115. PMC 4579343. PMID 26355004.
- ^ Hoeijmakers, Wieteke Anna Maria; Bártfai, Richárd (2018). "Characterization of the Nucleosome Landscape by Micrococcal Nuclease-Sequencing (MNase-seq)". Chromatin Immunoprecipitation. Methods in Molecular Biology. Vol. 1689. pp. 83–101. doi:10.1007/978-1-4939-7380-4_8. ISBN 978-1-4939-7379-8. ISSN 1064-3745. PMID 29027167.
{{cite book}}: Unknown parameter|name-list-format=ignored (|name-list-style=suggested) (help) - ^ a b c Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. (July 2015). "Single-cell chromatin accessibility reveals principles of regulatory variation". Nature. 523 (7561): 486–90. Bibcode:2015Natur.523..486B. doi:10.1038/nature14590. PMC 4685948. PMID 26083756.
- ^ a b Li, Zhijian; Schulz, Marcel H.; Zenke, Martin; Costa, Ivan G. (2018-07-08). "Identification of Transcription Factor Binding Sites using ATAC-seq". doi:10.1101/362863.
{{cite journal}}: Cite journal requires|journal=(help); Unknown parameter|name-list-format=ignored (|name-list-style=suggested) (help) - ^ Spektor R, Tippens ND, Mimoso CA, Soloway PD (June 2019). "methyl-ATAC-seq measures DNA methylation at accessible chromatin". Genome Research. 29 (6): 969–977. doi:10.1101/gr.245399.118. PMC 6581052. PMID 31160376.
- ^ Hendrickson, David G.; Soifer, Ilya; Wranik, Bernd J.; Botstein, David; Scott McIsaac, R. (2018), Simultaneous Profiling of DNA Accessibility and Gene Expression Dynamics with ATAC-Seq and RNA-Seq, Methods in Molecular Biology, vol. 1819, Springer New York, pp. 317–333, doi:10.1007/978-1-4939-8618-7_15, ISBN 9781493986170, PMID 30421411
{{citation}}: Unknown parameter|name-list-format=ignored (|name-list-style=suggested) (help) - ^ Prescott SL, Srinivasan R, Marchetto MC, Grishina I, Narvaiza I, Selleri L, et al. (September 2015). "Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest". Cell. 163 (1): 68–83. doi:10.1016/j.cell.2015.08.036. PMC 4848043. PMID 26365491.
- ^ Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, et al. (August 2014). "Immunogenetics. Chromatin state dynamics during blood formation". Science. 345 (6199): 943–9. doi:10.1126/science.1256271. PMC 4412442. PMID 25103404.
- ^ Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. (October 2018). "The chromatin accessibility landscape of primary human cancers". Science. 362 (6413): eaav1898. Bibcode:2018Sci...362.1898C. doi:10.1126/science.aav1898. PMC 6408149. PMID 30361341.
- ^ Wang J, Zibetti C, Shang P, Sripathi SR, Zhang P, Cano M, et al. (April 2018). "ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration". Nature Communications. 9 (1): 1364. Bibcode:2018NatCo...9.1364W. doi:10.1038/s41467-018-03856-y. PMC 5893535. PMID 29636475.
- ^ a b Mezger A, Klemm S, Mann I, Brower K, Mir A, Bostick M, et al. (September 2018). "High-throughput chromatin accessibility profiling at single-cell resolution". Nature Communications. 9 (1): 3647. Bibcode:2018NatCo...9.3647M. doi:10.1038/s41467-018-05887-x. PMC 6128862. PMID 30194434.
- ^ Cusanovich, Darren (May 2015). "Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing". Science. 348 (6237): 910–914. doi:10.1126/science.aab1601. PMC 4836442. PMID 25953818.
- ^ Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, Pokholok D, Aryee MJ, et al. (2019). "Droplet-based combinatorial indexing for massive scale single-cell epigenomics". bioRxiv. doi:10.1101/612713.
- ^ Chen X, Miragaia RJ, Natarajan KN, Teichmann SA (December 2018). "A rapid and robust method for single cell chromatin accessibility profiling". Nature Communications. 9 (1): 5345. Bibcode:2018NatCo...9.5345C. doi:10.1038/s41467-018-07771-0. PMC 6297232. PMID 30559361.
- ^ a b Li, Zhijian; Kuppe, Christoph; Cheng, Mingbo; Menzel, Sylvia; Zenke, Martin; Kramann, Rafael; et al. (2019-12-05). "scOpen: chromatin-accessibility estimation of single-cell ATAC data". bioRxiv: 865931. doi:10.1101/865931.
{{cite journal}}: Unknown parameter|name-list-format=ignored (|name-list-style=suggested) (help) - ^ Schep AN, Wu B, Buenrostro JD, Greenleaf WJ (October 2017). "chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data". Nature Methods. 14 (10): 975–978. doi:10.1038/nmeth.4401. PMC 5623146. PMID 28825706.