
Reverse pharmacology

Reverse Pharmacology or target-based pharmacology, is a process of drug development where identity of a molecular target (receptor, enzyme, protein, etc) drives compound screening. Classical pharmacology involves determining the functional activity of a compound through in vitro and in vivo models. Once the activity of compound is found, the compounds ligands are identified, purified, and synthesized and go through biological screening assays. The most selective and potent drug is then further screened for toxicity and efficacy. Classical pharmacology can be time consuming and expensive. Reverse pharmacology was first established in the 70's by Dir Ram Nath Chopra and Gannath Sen[1]. Reverse pharmacology, in contrast, takes potential drug compounds, designed specifically to targets (receptors, enzyme, proteins, etc) involved in disorders or diseases. Binding assays are used to identify the molecular target. The compound then undergoes animal functional studies to show the desired effect. Compounds identified through reverse pharmacology are thought to increase efficacy[2]. The goal of reverse pharmacology is to utilize disease pathology in order to identify specific and targetable elements that novel compounds can be modeled from.
Reverse Vaccinology
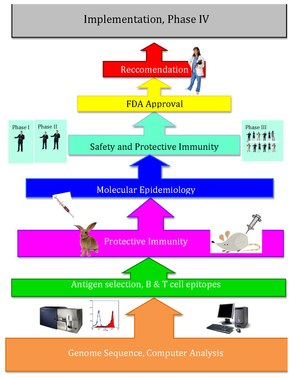
A sub category of reverse pharmacology, reverse vaccinology is a computational approach for discovery of vaccines through utilization of the genome. Traditionally vaccines have been developed through the isolation, inactivation, and re-injection of viruses. Conventional vaccinology is both time consuming and limited to antigens that are able to be purified for testing[3].
Rino Rappuoli and the J. Craig Venter Institute used reverse vaccinology to develop a vaccine against Serogroup B meningococcus.[4] Vaccines utilizing reverse vaccinology tend to have better selectivity; reducing side effects. These vaccines can increase immunity of multiple strains by incorporating multiple proteins.[5]
The reverse vaccinology approach uses the genome sequence of the pathogen itself. Researchers are able to determine the all of the protein antigens that a pathogen can express. Reverse vaccinology begins with the genomic sequence of the pathogen and computer prediction of canidates for vaccines. Scientists use computational analysis to obtain the genome of the virus which allows for the determination of proteins that are secreted during viral infection. Through the secreted proteins, scientists are then able to identify and purify the virus, allowing further research consisting of immunizing laboratory animals. The elicited immune response is studied and are used for identification of a vaccine. [6] Conventional vaccinology differs from reverse vaccinology in that the proteins purified from a cultured pathogen are used as candidates for a vaccine. [7]
Applications of Reverse Vaccinology
Diseases such as Malaria, Tuberculosis, and Syphilis have fully been sequences and lists of all possible genes can be accessed.
Group B Menigococcus
Group B Menigococcus is the first application of reverse vaccinology. The polysaccharide that was used to develop early vaccines was poorly immunogenic and caused autoimmunity. A vaccine needed to be made against the surface exposed proteins and were able to be folded within the outer membrane. Rino Rappuoli and the J. Craig Venter Insitute were able to screen DNA fragments for genes that coded surface exposed and exported proteins. These proteins were purified and used to immunize mice. 25 of 85 surface proteins were shown to produce antibodies. These proteins were the basis of the vaccine against Group BMenigococcus[3].
Limits to Reverse Vaccinology
The goal a vaccine is protective immunity against a pathogen. Vaccinology relies the availability of databases that can predict whether the candidates can provide protective immunity against the pathogen. Lack of knowledge surrounding vaccine immunology and effects of mutations down the line, it is hard to predict protective immunity. Another limitation of reverse vaccinology is the identification antigens that are not proteins.
Reverse Vaccinology Tools and Applications
More than 4000 viral genomes have been identified.[6] Reverse vaccinology heavily uses bioinformatics to analyze and obtain vast viral genomes.
NERVE
New Enhanced Reverse Vaccinology Environment (NERVE) is a reverse vaccinology software that imports pathogen protein sequences and predicts biological sequences. This software predicts the sub-cellular localization, adhesion probability, topology, human sequence similarity, and conservation of these proteins. NERVE uses four criteria to predict potential vaccine candidates: proteins that do not lie in the cytoplasm, proteins with 2 or less transmembrane helices, a probability of adhesion >0.46 and proteins that have low similarity to human proteins[8].
Vaxign
Vaxign was the first vaccine design program for reverse vaccinology and vaccine development. It uses both external and internal tools and programs to predict vaccine targets. Users input amino acids from proteins are genomes and is able to predict subcellular localization, transmembrane domains, adhesion probability, protein conservation among genomes, exclusion of nonpathogenic strains, comparison of proteins and host, prediction of binding to MHC class I and II, and analysis of the protein function.[9]
Vaxign has two broad methods of vaccine design: "General" and "Specific" Methods. Within "General Methods" users can further choose to search under Vaxign Query or Dynamic Vaxign Analysis. Vaxign Query allows the users to search precomputed results for around 300 genomes. Users are able to choose genomes for vaccine targets based on desired parameters or protein sequences. The Dynamic Vaxign Analysis has users input protein sequences and set up parameters. This analytical tool uses the automatic Vaxign pipeline[10]. This pipeline includes predictions for sublocation, adhesion, epitope binding to MCH class I and class II, and similarity to the host genome sequences.
Under the "Specific Methods", users have the option of Vaxitop and Vaxign-ML. Vaxitop makes predictions on vaccine epitopes based on reverse vaccinology[11]. Vaxitop specifically predicts the binding to MHC Class and II. Vaxitop allows users to perform a genome whide query for different MHC host species. Vaxign-ML uses machine learning to produce vaccine candidates[12].
EpiVax and iVAX Toolkit
EpiVax is a private company, based in Providence, RI, that uses in silico, in vitro, and in vivo applications to design new vaccines. EpiVax created the iVAX Toolkit, an in silico platform that allows users to identify and predict epitopes for vaccine development.[13]
iVAX is a computational vaccine design program that encompasses epitope mapping, antigen selection, and immunogen design. This toolkit uses immunoinformatics algorithms to identify candidate antigens and select for conserved T cell epitopes, eliminating epitopes from regulatory T cells. iVAX has a collection of tools such as Conservatrix, EpiMatrix, ClustiMer, and EpiAssembler. Vaccine design begins with searching for MHC class I and II ligands. EpiMatrix performs this initial search by parsing and evaluating each input sequence for binding efficacy. The program removes low quality binders to curate personalized predictions. These epitopes can be further analyzed for clusters using ClustiMer. Users can find cross-strain, conserved epitopes using Conservatrix[14]. This toolkit integrates in silico and ex vivo/ in vitro technology to allow vaccine developers to access toxicity, efficacy, and performance of vaccines.
Reverse Pharmacognosy
Pharmacognosy is a multi disciplinary science that studies the applications of natural compounds. Pharmacognosy is derived from the Greek pharmakon, meaning drug or recipe, and gnosis, meaning knowledge. Pharmacognosy is not limited to the natural compound application in therapeutics, but can also include cosmetics, agricultures, and dyes. Conventional pharmacognosy utilizes traditional knowledge of living organisms to find new bioactive molecules. Conventional pharmacognosy begins with using ethopharmacological data to select plants. Once these plants are selected, extracts of these plants are made and tested in biological assays. If an extract is biologically active, the extracts are fractionated and retested multiple times to identify the molecules responsible for the activity.
Reverse Pharmacognosy attempts to use the knowledge generated from pharmacognosy to introduce new therapeutic activities of natural products. Molecules are first selected based on criteria (eg. structure, chemical family, activity). Next, the selected compounds are used to identify potential targets. Compounds can have a variety of different targets in metabolic pathways. This information gives insight on potential off-target effects and synergetic applications. "Inverse screening" involves identifying new properties for the selected compounds. Predicted interaction partners can be validated using in vitro binding assays or virtual screenings[15].
Summary of Reverse Pharmacognosy[16]
Selection of Molecules
The first step in reverse pharmacognosy is the selection of natural compounds. Criteria can be applied depending on the compound is proposed for: structural criteria, molecules from the same chemical family, compounds with drug-like properties, etc. Natural product databases can also be helpful for compound selection.
Target Identification and Discovery of Activities
The second step in reverse pharmacognosy is identifying the target which will bind to the selected compounds. There can be many targets that a ligand can interact with, these interactions can illicit either negative or favorable effects, so it is important to identify all possible interactions. Researchers at this step commonly use "inverse screening" where they screen proteins which will potentially bind their molecules. Predictions about selectivity and synergy can be calculated which cannot be achieved through classical docking.
Biological Assays and Organism Associated Activities
While virtual screening are fairly accurate at predicting the biological activities of compounds and their proteins, these interactions can only be confirmed through in vitro biological assays. in vivo models of biological activities are needed to confirm that there is the same biological properties from the in vitro experiments.
Activity Optimization
Derivatives of natural products may be more potent, less toxic, more accessible from the compounds that were originally probed. Database of active extracts and metabolites can assist with this optimization.
Reverse Pharmacognosy Tools and Applications:
Inverse Screening Tools and Target Databases
| Database Name | Information | Website |
|---|---|---|
| Dr. Duke's Database | Contains 50,000 entries on plants, chemicals, biological activities, syndrome, ethnobotany plants, and ethnobotany use | [1] |
| ChemNetBase | Collection of Interactive Databases and Dictionaries:
-Combined Chemical Dictionary -Dictionary of Natural Products Dictionary of Organic Compounds -Dictionary of Drugs -Dictionary of Inorganic and Organometallic Compounds -Dictionary of Commonly Cited Compounds -Dictionary of Marine Natural Products -Dictionary of Food Compounds -Polymers: A Property Database -Propertied of Organic Compounds |
[2] |
| Napralert | Relational database of natural products. Contains enthnomedical, pharmacological, and biochemical information on natural products | [3] |
| Plants for a Future (PFAF) | Contains the edible and medicinal uses of 7000+ plant. | [4] |
| Supernatural | Contains 325,000+ natural products and their structures and physiochemical properties. | [5] |
Greenpharma's Reverse Pharmacognosy Platform
Greenpharma is a French R&D company created in 2000 who supplies tailored products and services in the life sciences. They focus on natural substances and their platform consists of five components: analytical chemistry, lab scale extractions, chemoinformatics, organic/bio synthesis, and cosmetic formulations. Greenpharma offers three compound libraries for reverse pharmacognosy needs: Greenpharma Natural Compound Library (GPNCL), Greenpharma Ligand Library (LIGENDO), and Greenpharma Plant Extract Library (GPEL)[17].
The GPNCL is a collection of 150,000 natural compound structures for lead discovery. This library also has access to 30 million compounds from Ambinter. This library does not include amino acids, peptides, nucleic acids, or long fatty acid chains. They also have continuous stock of compounds at >90% purity. The GPNCL provides the physico-chemical properties and phytochemistry of each of their compounds for researchers[17].
LIGENDO is a library source of natural, pure compounds for chemogenomics and biological pathway hopping. This library is composed of 400 human endogenous ligands. Compounds are given in microplates of 80 and data is supplied in the database with compound name, structure, implied metabolic pathway, physico-chemical properties, and protein partners[17].
GPEL is a plant extract library that combines botany, pharmacology, and pharmacognosy to present a wide range of possible extracts. The library is suitable for a high throughput screening which 80 extracts on each microplate for 20 plants. Greenpharma provides 4 different polarity solvent fractions. Information on the plant family, genus, species, and organ data is also provided[17].
Selnergy
Selnergy is a virtual high throughput screening platform that allows users to explore interactions chemogenomics. It contains a database of 10,000 protein structures, sectioned by their biological properties. This platforms allows for in silico profiling of ligand and ligand-protein interactions. It allows the user to predict the selectivity and synergy between candidate compounds and their protein targets[18].
Potential Use in Traditional Systems of Medicine
Current drug discovery entails the identification of drug targets in disease pathology, large iterations of chemical compounds to discover drug candidates and performing biological assays to test for toxicity, potency, and efficacy. This traditional approach is often considered costly and time consuming. Much of the world relies on traditional systems of medicine (TSM): Ayurveda, traditional Chinese medicine (TCM), etc. While these therapies are popular in non-western countries, their evidence of therapeutic benefits are seen as incomplete in western societies. Reverse pharmacology began as a way to study Ayurvedic plants chemically and clinically. The issue with studying Ayurvedic plants is that there was no defined approach in quantifying its benefits. The study of their herbal therapies can be investigated using reverse pharmacology.
Target Identification and Characterization
Proteins that are thought to be critical in pathogenesis are identified. These protein structures and their predicted function can be analyzed through bioinformatics. This enables the identification of candidate ligands. Receptor/ ligand interactions are often identified through high throughput screenings.[19]

High Throughput Screening
High throughput screening is method in drug discovery that allows scientists to conduct pharmacological tests. In regards to reverse pharmacology, this process is utilized for the identification of compounds involved in a pathophysiology pathway.

Molecular Docking
Molecular docking is an in silico method used in drug discovery to identify novel compounds of interest. It has the ability to predict the binding conformation of small molecule ligands to their binding site. Docking was first introduced in the 1970s, and allows researchers the ability to predict interactions between the target and potential ligands.[20]
Molecular docking is currently used for the prediction of targets for compounds, prediction of adverse drug reactions, polypharmaoclogy, virtual screenings, and drug repositioning. Virtual screening using molecular docking utilizes large collections of synthesized and designed molecules to find macromolecule binding sites. Curated molecules are scored based on their binding energies and other parameters[21].
Ligand-Based approaches are used to identify suitable protein conformations for the docking screenings. This approach can also be used to confirm the prediction from the docking screenings. Researchers can use the similarity between the predicted binding confirmation and the experimental conformation when the ligand is crystalized with the protein. Molecular dynamics (MD) and binding free energy estimations are both structure based approaches, often used in combination. Residue flexibility and conformational changes can be evaluated through molecular dynamics. It can be used to determine the stability of different protein conformations. The use of artificial intelligence and statistical methods are new in the molecular docking pipeline[20]. These methods can utilize publicly available information on the structural, chemical and activity of compounds for better predictions.
Reverse Docking

Reverse docking, or inverse docking, is an in silico method to find proteins targets to a specific ligand[22]. Much like regular docking methods, ligand-target conformations are scored and ranked based on preset parameters[23]. Large and properly constructed databases need to be created with target structures. These databases also need to define the binding sites for each proteins. There are multiple reverse docking tools that use a variety of databases and can be used in identifying targets and potential off-target interactions. The problem with efficacy of reverse docking comes from high computational time and lack of databases for target structures[24].
Reverse Docking Programs
INVDOCK
INVDOCK was the first reverse docking tool, created in 2001. This software aligns ligands to the binding site and binding conformations are analyzed using energy minimization. INVDOCK can be used to identify unknown and secondary targets of drugs, leads, etc. It can also predict the ADME of targets[25]. One of the limitations of INVDOCK is the lack of optimization; users input a threshold binding energy and once the ligand is positioned successfully, within the binding energy threshold, the program moves on. There is no optimization for multiple low energy conformations.
TarFisDock
TarFisDock was first developed in 2006 and is a tool that ranks targets using an in house database, Potential Drug Target Database (PDTD)[26]. This tool calculates the binding energy of targets and their ligands. Users input their small molecule to be tested and TarFisDock searches for proteins using docking techniques. Viable targets are usually contained in the top 2,5, or 10% of its rankings.
idTarget
idTarget is a web based docking tool that allows for multiple binding sites of a protein to be identified[26]. This tool uses all the protein structures within the Protein Data Bank (PDB). This tool is also able to determine off targets of compounds.
References
- ^ Blondeau, S.; Do, Q.T.; Scior, T.; Bernard, P.; Morin-Allory, L. (2010-05-01). "Reverse Pharmacognosy: Another Way to Harness the Generosity of Nature". Current Pharmaceutical Design. 16 (15): 1682–1696. doi:10.2174/138161210791164036. ISSN 1381-6128.
- ^ Shaikh, Mohd Farooq (2016-05-24). "Reverse Pharmacology: Fast Track Path of Drug Discovery". Pharmacy & Pharmacology International Journal. 4 (3). doi:10.15406/ppij.2016.04.00077.
- ^ a b Rappuoli, Rino (2000). "Reverse vaccinology". Current Opinion in Microbiology. 3: 445–450 – via Elseiver Science Ltd.
- ^ Sette, Alessandro; Rappuoli, Rino (2010-10). "Reverse Vaccinology: Developing Vaccines in the Era of Genomics". Immunity. 33 (4): 530–541. doi:10.1016/j.immuni.2010.09.017. PMC 3320742. PMID 21029963.
{{cite journal}}: Check date values in:|date=(help)CS1 maint: PMC format (link) - ^ Hansson, Marianne; Nygren, Per-Åke; Ståhl, Stefan (2000-10-01). "Design and production of recombinant subunit vaccines". Biotechnology and Applied Biochemistry. 32 (2): 95. doi:10.1042/ba20000034. ISSN 0885-4513.
- ^ a b Bruno, Luca; Cortese, Mirko; Rappuoli, Rino; Merola, Marcello (2015-04-01). "Lessons from Reverse Vaccinology for viral vaccine design". Current Opinion in Virology. Viral pathogenesis • Preventive and therapeutic vaccines. 11: 89–97. doi:10.1016/j.coviro.2015.03.001. ISSN 1879-6257.
- ^ Heinson, A. I.; Woelk, C. H.; Newell, M.-L. (2015-03-01). "The promise of reverse vaccinology". International Health. 7 (2): 85–89. doi:10.1093/inthealth/ihv002. ISSN 1876-3413.
- ^ Dalsass, Mattia; Brozzi, Alessandro; Medini, Duccio; Rappuoli, Rino (2019-02-14). "Comparison of Open-Source Reverse Vaccinology Programs for Bacterial Vaccine Antigen Discovery". Frontiers in Immunology. 10. doi:10.3389/fimmu.2019.00113. ISSN 1664-3224.
{{cite journal}}: CS1 maint: unflagged free DOI (link) - ^ He, Yongqun; Xiang, Zuoshuang; Mobley, Harry L. T. (2010). "Vaxign: The First Web-Based Vaccine Design Program for Reverse Vaccinology and Applications for Vaccine Development". Journal of Biomedicine and Biotechnology. 2010: 1–15. doi:10.1155/2010/297505. ISSN 1110-7243. PMC 2910479. PMID 20671958.
{{cite journal}}: CS1 maint: PMC format (link) CS1 maint: unflagged free DOI (link) - ^ Xiang, Z; He, Y (2009). "Vaxign: a web-based vaccine target design program for reverse vaccinology". Procedia in Vaccinology. 1: 23–29.
- ^ He, Yongqun; Xiang, Zuoshuang; Mobley, Harry L. T. (2010). "Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development". Journal of Biomedicine & Biotechnology. 2010: 297505. doi:10.1155/2010/297505. ISSN 1110-7251. PMC 2910479. PMID 20671958.
{{cite journal}}: CS1 maint: unflagged free DOI (link) - ^ Ong, Edison; Wang, Haihe; Wong, Mei U.; Seetharaman, Meenakshi; Valdez, Ninotchka; He, Yongqun (2020-05-01). "Vaxign-ML: supervised machine learning reverse vaccinology model for improved prediction of bacterial protective antigens". Bioinformatics (Oxford, England). 36 (10): 3185–3191. doi:10.1093/bioinformatics/btaa119. ISSN 1367-4811. PMC 7214037. PMID 32096826.
- ^ De Groot, Anne S.; Jesdale, Bill M.; Szu, Evan; Schafer, James R.; Chicz, Roman M.; Deocampo, Greg (1997-05). "An Interactive Web Site Providing Major Histocompatibility Ligand Predictions: Application to HIV Research". AIDS Research and Human Retroviruses. 13 (7): 529–531. doi:10.1089/aid.1997.13.529. ISSN 0889-2229.
{{cite journal}}: Check date values in:|date=(help) - ^ Moise, Leonard; Gutierrez, Andres; Kibria, Farzana; Martin, Rebecca; Tassone, Ryan; Liu, Rui; Terry, Frances; Martin, Bill; De Groot, Anne S (2015-09-02). "iVAX: An integrated toolkit for the selection and optimization of antigens and the design of epitope-driven vaccines". Human Vaccines & Immunotherapeutics. 11 (9): 2312–2321. doi:10.1080/21645515.2015.1061159. ISSN 2164-5515. PMC 4635942. PMID 26155959.
{{cite journal}}: CS1 maint: PMC format (link) - ^ Do, Quoc-Tuan; Bernard, Philippe (2006), "Reverse pharmacognosy: a new concept for accelerating natural drug discovery", Lead Molecules from Natural Products - Discovery and New Trends, Elsevier, pp. 1–20, retrieved 2021-11-28
- ^ Blondeau, S.; Do, Q.T.; Scior, T.; Bernard, P.; Morin-Allory, L. (2010-05-01). "Reverse Pharmacognosy: Another Way to Harness the Generosity of Nature". Current Pharmaceutical Design. 16 (15): 1682–1696. doi:10.2174/138161210791164036. ISSN 1381-6128.
- ^ a b c d "Compound Librairies". Greenpharma. Retrieved 2021-11-27.
- ^ "Drug discovery". Greenpharma. Retrieved 2021-11-28.
- ^ Takenaka, T. (2001-09). "Classical vs reverse pharmacology in drug discovery: CLASSICAL VS REVERSE PHARMACOLOGY". BJU International. 88: 7–10. doi:10.1111/j.1464-410X.2001.00112.x.
{{cite journal}}: Check date values in:|date=(help) - ^ a b Pinzi, Luca; Rastelli, Giulio (2019-09-04). "Molecular Docking: Shifting Paradigms in Drug Discovery". International Journal of Molecular Sciences. 20 (18): 4331. doi:10.3390/ijms20184331. ISSN 1422-0067. PMC 6769923. PMID 31487867.
{{cite journal}}: CS1 maint: PMC format (link) CS1 maint: unflagged free DOI (link) - ^ Kumar, Shivani; Kumar, Suresh (2019), "Molecular Docking: A Structure-Based Approach for Drug Repurposing", In Silico Drug Design, Elsevier, pp. 161–189, retrieved 2021-11-28
- ^ Pinzi, Luca; Rastelli, Giulio (2019-09-04). "Molecular Docking: Shifting Paradigms in Drug Discovery". International Journal of Molecular Sciences. 20 (18): 4331. doi:10.3390/ijms20184331. ISSN 1422-0067. PMC 6769923. PMID 31487867.
{{cite journal}}: CS1 maint: PMC format (link) CS1 maint: unflagged free DOI (link) - ^ Kharkar, Prashant S; Warrier, Sona; Gaud, Ram S (2014-03). "Reverse docking: a powerful tool for drug repositioning and drug rescue". Future Medicinal Chemistry. 6 (3): 333–342. doi:10.4155/fmc.13.207. ISSN 1756-8919.
{{cite journal}}: Check date values in:|date=(help) - ^ Lee, Aeri; Lee, Kyoungyeul; Kim, Dongsup (2016-07-02). "Using reverse docking for target identification and its applications for drug discovery". Expert Opinion on Drug Discovery. 11 (7): 707–715. doi:10.1080/17460441.2016.1190706. ISSN 1746-0441. PMID 27186904.
- ^ "Bioinformatics & Drug Design group [BIDD]". bidd.group. Retrieved 2021-11-28.
- ^ a b Lee, Aeri; Lee, Kyoungyeul; Kim, Dongsup (2016-06). "Using reverse docking for target identification and its applications for drug discovery". Expert Opinion on Drug Discovery. 11 (7): 707–715. doi:10.1080/17460441.2016.1190706. ISSN 1746-0441.
{{cite journal}}: Check date values in:|date=(help)
This pharmacology-related article is a stub. You can help Wikipedia by expanding it. |