
Complejo (química)

2(NH
3)
2
Un complejo formado por un átomo de platino coordinado con dos cloruros (en verde) y dos grupos amino. Este complejo basado en el platino reacciona in vivo, uniéndose al ADN celular y causando apoptosis, por lo que se utiliza como agente quimioterápico en el tratamiento de muchos tipos de cáncer.


En química se denomina complejo a una entidad que se encuentra formada por una asociación que involucra a dos o más componentes unidos por un tipo de enlace químico, el enlace de coordinación, que normalmente es un poco más débil que un enlace covalente típico.[1]
Por una costumbre histórica el término complejo se utiliza principalmente para describir a aquel tipo de estructura molecular que usualmente se encuentra formada por un átomo central (el cual es con frecuencia un catión metálico) que se encuentra enlazado a otras entidades moleculares que lo rodean llamadas ligandos. Esta última acepción también se conoce como entidad de coordinación.[2]
El término también es utilizado para referirse a una enorme cantidad de estructuras inestables o metaestables que participan como intermediarias en diferentes reacciones; por lo cual es preferible utilizar siempre que se pueda un término más explicativo para referirse a estos compuestos. En este sentido el término complejo es mucho más amplio, pero menos preciso. En química inorgánica, por ejemplo, se prefiere utilizar el término entidad de coordinación en lugar de complejo.
La química de los complejos tiene numerosas aplicaciones tanto teóricas como prácticas sirviendo por ejemplo: para explicar detalles tan comunes como el color de las piedras preciosas; la elaboración industrial de polímeros, pigmentos, vidrios incoloros y de colores; electrodepósito de metales; formulación de ablandadores de agua para productos de limpieza hogareños y hasta el tratamiento de algunas intoxicaciones y la base teórica que permite comprender la mayoría de las reacciones enzimáticas que permiten la existencia de la vida.
Consideraciones previas
[editar]Los complejos más sencillos responden a un tipo de estructura molecular que se encuentra generalmente formada por un grupo central (generalmente un catión) llamado núcleo de coordinación que posee orbitales de valencia no ocupados, rodeado por un cierto número de moléculas o iones que poseen pares de electrones no compartidos. Estos electrones no compartidos pueden ser inyectados en los orbitales vacíos del grupo central para formar enlaces coordinados.
Aunque en general el grupo central es un catión también puede ser un átomo neutro, por ejemplo un átomo de gas noble, o una molécula y puede poseer carga positiva, negativa o carecer por completo de carga.
A los iones o moléculas que participan de la estructura molecular inyectando su par de electrones no compartidos se les denomina genéricamente ligandos.
Al aducto formado por el grupo central y los ligandos se le denomina entidad de coordinación y a los compuestos que contienen entidades de coordinación en su constitución se les denomina compuestos de coordinación.
Un ligando enlazado a un átomo central se dice que está coordinado a ese átomo. El número de pares de electrones que es capaz de aceptar el grupo central se denomina número de coordinación.
Generalidades
[editar]Los átomos de los elementos metálicos tienen una clara tendencia a perder electrones para convertirse en iones con carga positiva (cationes), esto es así porque en general poseen un radio atómico elevado en relación con la carga de sus núcleos, lo que posibilita que sus electrones de valencia se desprendan con mucha facilidad. (Al ser los electrones de valencia los que se encuentran a mayor distancia del núcleo, son los que menos atracción electrostática experimentan y por lo tanto son los que se desprenden con mayor facilidad.)
Esto puede conducir a la idea de que los iones metálicos con carga positiva (cationes) deberían ser muy abundantes en la naturaleza. Sin embargo los cationes metálicos rara vez se encuentran en estado libre en la naturaleza, esto es así porque al perder uno o más electrones su radio disminuye y su carga eléctrica aumenta. Un aumento en la relación carga/radio significa una disminución de la estabilidad termodinámica de una especie química.
En general los cationes metálicos poseen una relación carga/radio tan elevada que rápidamente interactúan con otros iones, átomos o moléculas, para adquirir una estructura que resulte termodinámicamente más estable. A esta estabilización la consiguen ya sea interactuando con moléculas neutras, lo que provoca un aumento del radio molecular y una consiguiente disminución de la relación carga/radio; o con iones de con carga negativa (aniones) los que además de provocar un aumento en el radio molecular brindan una estabilidad adicional al “aliviar” al catión aportando cargas negativas.
Es común que en estas asociaciones, las moléculas o iones que otorgan estabilidad al catión central actúen como bases de Lewis, es decir, que al poseer uno o más pares de electrones no compartidos sean capaces de "inyectar" esos electrones en orbitales vacíos del catión para aumentar su estabilidad.
Los cationes metálicos casi siempre se encuentran en la naturaleza formando algún tipo de complejo que los estabiliza; con mucha frecuencia el agente acomplejante suele ser el solvente donde se encuentran disueltos.
Una buena parte de las sales metálicas de los metales de los grupos principales y de transición se encuentran hidratadas. Las aguas de hidratación actúan como ligandos que rodean al metal, enlazándose a través de un par electrónico no compartido del agua. Un ejemplo notable de esto son las sales de cobalto que se utilizan para "predecir el tiempo" en algunos juegos infantiles, en estas el cobalto se encuentra coordinado por un número de moléculas de agua que cambia con la humedad ambiental, el cambio en la coordinación del cobalto provoca un cambio en el color de la sal, de azul a rosado al aumentar la humedad y a la inversa.
La mayor parte de la química de complejos conocida trata de complejos formados por entidades de coordinación con núcleos de coordinación que son cationes metálicos, sin embargo debe quedar claro que también existen complejos en los cuales estos núcleos de coordinación son átomos no metálicos, o participan con carga cero, o se trata de moléculas en lugar de átomos. Incluso hasta hay algunos en los cuales el núcleo tiene carga negativa.
Historia
[editar]
Con el constante avance que tuvo la química en sus comienzos como ciencia exacta, pronto se sintetizaron una serie de nuevos compuestos que resultaron muy llamativos, especialmente por sus colores. Estos compuestos químicos, a falta de una descripción más adecuada, tomaron los nombres de sus creadores. Así se dieron a conocer por ejemplo la sal de Magnus (2PtCl
2·2NH
3) o la sal de Erdmann (KNO
2·Co(NO
2)
2·2NH
2). Otro de estos vistosos compuestos fue el azul de Prusia, también conocido como Berliner Blau o azul de Berlín, (KCN·Fe(CN)
2·Fe(CN)
3) producido por Diesbach en Berlín a comienzos del siglo XVIII. Muchos de los primeros complejos fueron utilizados como pigmentos por los pintores de la época.
Estos compuestos presentaban dos notables propiedades que los diferenciaban de los conocidos hasta el momento: Primero los brillantes cambios de color asociados a su formación, y segundo la reactividad alterada de los iones que participaban.
Hasta mediados del siglo XIX los químicos no comenzaron a interesarse por la verdadera naturaleza de su constitución y por su relación con otros compuestos más sencillos.
Al principio se encontró, como se puede notar en las fórmulas arriba expresadas, que estos compuestos parecían estar formados por la asociación de otros compuestos más sencillos. Esto llevó a identificarlos como "compuestos moleculares", para diferenciarlos de los "compuestos atómicos" más simples; y por último, se les dio el nombre de COMPLEJOS, para diferenciarlos de los compuestos simples. A decir verdad, este era un nombre acertado para la época, pues era difícil encontrarles una estructura valedera que pudiera explicar todas sus propiedades. A la par de ello el número de complejos conocidos aumentaba a medida que progresaba su estudio.
El desarrollo de modelos que permitieran explicar su naturaleza, tuvo que esperar la aparición de teorías y modelos válidos para compuestos más sencillos. Los pasos fundamentales en este sentido fueron en primer lugar, la definición de lo que se consideraba un compuesto "verdadero", más conocida como Ley de las proporciones definidas, propuesta por J.L. Proust en 1799, la cual establece que: «un compuesto determinado siempre estará constituido por las mismas proporciones de sus elementos constituyentes». En otras palabras lo que hoy conocemos como "estequiometría definida". Esta primera definición fue trascendental para separar unos compuestos de otros, especialmente en el caso de los complejos.
Luego, en 1827, J.J. Berzelius introdujo el concepto de isomería, el cual complementa la definición anterior pero introduce una pregunta clave: ¿cómo se unen entre sí los átomos? Para encontrar una respuesta a esta pregunta fue definitiva la teoría de los tipos, propuesta por Ch. Gerhardt (hacia el año 1853), desarrollada para el amoníaco por Ch. A. Wurtz en 1849 y ampliada y trabajada por A. W. Von Hofmann (), hoy conocida como "Teoría del amonio"; esta teoría fue una primera aproximación para explicar cómo estaban unidos los átomos en los abundantes "complejos" que contenían amoníaco.
Simultáneamente, se desarrolla la "Teoría de la Fuerza de Combinación" o "Teoría de la atomicidad" (una primera aproximación al actual concepto de valencia, propuesta por E. Frankland en 1852 como una extensión de la ley de las proporciones definidas; esta teoría establece que «cada elemento sólo se puede unir a un número fijo de otros elementos». Así, se podía asegurar que la atomicidad del Zinc siempre era dos y la del nitrógeno o la del fósforo era 3 o 5.

F.A. Kekulé en 1858, propone la noción de que muchos compuestos orgánicos tenían que ser producto de la unión entre sí de átomos de carbono en forma de cadenas. Además de lo conocido con anterioridad, esta noción influyó necesariamente en un segundo modelo desarrollado en 1869 y sustentado con éxito durante varias décadas por los escandinavos Blomstrand, profesor de Química en Lund (Suecia) y su alumno Jörgensen, quien fuera más tarde profesor en la Universidad de Copenhague y uno de los experimentadores más sobresalientes de la química de la coordinación. Sin embargo el mismo Jörgensen habría de sintetizar el compuesto que definiría la validez de su teoría, concluyendo al final que su modelo, conocido como teoría de las cadenas, era incorrecto.

El reconocimiento de la verdadera naturaleza de los complejos, se inicia con Alfred Werner (1866-1919) profesor de Química en Zúrich, quien demostró que las moléculas neutras que participaban en la formación de un complejo (entidad de coordinación) estaban directamente enlazadas al metal, de manera tal que las sales complejas como el CoCl
3·6NH
3 debían ser formuladas correctamente como [Co(NH
3)
6]3+
Cl−
3. También demostró que se originaban profundas consecuencias estereoquímicas si se hacía la suposición de que las moléculas o iones (ligandos) alrededor del metal ocupaban posiciones en los vértices de un cuadrado o de un octaedro.
Alfred Werner, propuso que los átomos podían exhibir simultáneamente más de un tipo de valencia. La primera parte de su teoría de la coordinación, publicada en 1893, puede resumirse en los siguientes tres postulados:
- 1. La mayoría de los elementos químicos presentan dos tipos de valencia, la valencia primaria o unión ionizable, hoy conocida como número de oxidación y la valencia secundaria o unión no ionizable, hoy conocida como número de coordinación.
- 2. Los elementos tienden a satisfacer tanto su valencia primaria como su valencia secundaria.
- 3. La valencia secundaria o número de coordinación, está distribuida en posiciones definidas en el espacio.
Estos tres postulados daban una explicación satisfactoria a las principales preguntas que se venían planteando los químicos con respecto a los "complejos". Tomando como ejemplo para la aplicación de los postulados la bien conocida serie de las aminas CoCl
3·6NH
3, CoCl
3·5NH
3, CoCl
3·4NH
3, CoCl
3·3NH
3, se tiene que la valencia primaria o estado de oxidación del cobalto en todos los casos es 3+, y la valencia secundaria o número de coordinación de este ion es 6. El estado de oxidación 3+ del cobalto está compensado, como se ve claramente en todos los casos, por 3 iones cloruro. En el primer ejemplo todos los cloruros son iónicos y no forman parte del catión complejo [Co(NH
3)
6]3+
; el número de coordinación 6 está satisfecho por 6 grupos NH
3. En el segundo ejemplo, el número de coordinación está satisfecho por 5 NH
3 y 1 Cl−
, [CoCl(NH
3)
5]2+
y únicamente dos cloruros son iónicos. En el tercer caso, 4 NH
3 y 2 Cl−
satisfacen el número de coordinación [CoCl
2(NH
3)
4]+
y en el último caso lo satisfacen 3 NH
3 y 3 Cl−
, [CoCl
3(NH
3)
3].
El tercer postulado llevó a Werner a afirmar que la presencia de isomería óptica para complejos del tipo [M(AA)
3] (Donde AA es un ligando bidentado) era evidencia de una estructura octaédrica y que esta isomería era debida a la asimetría de la molécula; algunos químicos orgánicos trataron de refutar su hipótesis aduciendo que la actividad óptica se debía a la presencia de átomos de carbono en la estructura y que esta propiedad era exclusivamente debida al carbono. Esta controversia llevó a Werner y su grupo en 1914 a sintetizar el más extraordinario complejo de la época, el hexol ([Co(Co(NH
3)
4)
3(OH)
6]), que no contenía carbono en su estructura.

El hexol presentó, como hoy es obvio, isomería óptica, consolidando la teoría de la coordinación, y además mostrando que esta isomería es una función de las operaciones de simetría de las moléculas en general y no específica de un único tipo de átomo.
La conclusión de Werner sentó uno de los argumentos de mayor peso para considerar a la química como una sola, con normas generales, y derrumbar murallas impuestas artificialmente entre la química orgánica y la inorgánica.
Werner obtuvo el Premio Nobel en 1913 por el desarrollo de su teoría de la coordinación. En sus conceptos fundamentales, esta teoría continúa vigente ya que permite explicar correctamente muchos de los aspectos estructurales de los compuestos de coordinación.
Los estudios estereoquímicos de Werner fueron seguidos más tarde por las ideas de G. N. Lewis y N. V. Sidgwick, quienes propusieron que eran los electrones de la última órbita de un átomo los responsables de los enlaces químicos y que «un enlace químico requería compartir un par de electrones» de manera tal que quedara cumplida la condición de que cada átomo participante en el enlace obtuviera al final ocho electrones en su capa más externa (Regla del octeto).
Con respecto a los compuestos de coordinación, Lewis postuló que: «los grupos que están unidos al ion metálico, conformando la entidad de coordinación, poseen pares libres de electrones, es decir, que no están compartidos en un enlace» y definió el número de coordinación como «el número real de pares de electrones que están unidos al átomo metálico».
En otro aspecto de su teoría, Lewis propuso una definición más general para ácidos y bases, en la cual una base es aquella que tiene un par libre de electrones que puede donar a otro átomo, mientras que un ácido es la sustancia que puede aceptar un par libre de electrones para formar un enlace. En este sentido, el ion metálico en un complejo es un ácido de Lewis y los grupos que están unidos a este ion en la entidad de coordinación son bases de Lewis.
Lewis propuso su modelo en 1916 y Sidgwick lo amplió hacia 1927, y resultó una verdadera revolución en la química porque permitió explicar de manera sencilla la naturaleza del enlace químico en compuestos sumamente diversos, llegando por ejemplo a considerar bajo esta óptica, a toda la química de complejos como simples reacciones ácido-base.
EL modelo de Lewis fue posteriormente ampliado y completado por la Teoría del Enlace de Valencia (TEV) y la Teoría de Orbitales Moleculares (TOM) que nos permiten actualmente interpretar la gran mayoría de las reacciones y propiedades de los complejos.

Naturaleza de la unión grupo central-ligando
[editar]Normalmente los ligandos son nucleófilos, aniones, moléculas polares o fácilmente polarizables que poseen al menos un par de electrones de valencia no compartidos, tales como H
2O, NH
3, X−
, RCN−
, etc.
Esto induce en un principio a tratar de explicar de manera sencilla las atracciones que se establecen entre ligandos y cationes como de naturaleza electrostática: el par de electrones del ligando es intensamente atraído por la alta carga del catión, forzando a la molécula o anión que lo posee a acercarse.
Sin embargo este enfoque no permite explicar cómo se forman los complejos con grupos centrales neutros o con carga negativa.
Una mejor aproximación es considerar a la unión grupo central-ligando como un tipo particular de aducto de Lewis en el cual participan los electrones del par electrónico no compartido del ligando y los orbitales vacíos (ya sean atómicos o moleculares) del grupo central. En este enlace el ligando aporta un par de electrones de valencia no compartidos (base de Lewis), y el grupo central los acepta (ácido de Lewis) para formar uno de los enlaces covalentes del complejo.
La unión que se establece entre grupo central y ligando es por tanto de tipo covalente.
Este tipo de unión covalente en el cual uno de los átomos aporta los dos electrones del enlace, recibe el nombre de enlace covalente coordinado.
Con base en este modelo, algunos autores hacen una diferencia entre el enlace covalente propiamente dicho, en donde se supone que cada átomo comprometido aporta un electrón para formar el enlace por par electrónico, y el enlace de coordinación, en donde se propone que solo uno de los átomos comprometidos en el enlace aporta el par de electrones. Si bien esta diferenciación ayuda a entender el origen del enlace, una vez formado el compuesto de coordinación ya no tiene sentido, puesto que los enlaces son equivalentes.
El ejemplo más sencillo para ilustrar lo anterior es el del H
3N y el H
3N-H+
o mejor NH+
4, en el que se podría pensar que este último enlace es diferente por ser coordinado y en algunos textos hasta se llega a representar como H
3N→H+
; sin embargo, el ion NH+
4 es un tetraedro regular, en el que cada uno los cuatro enlaces es equivalente a los otros y por lo tanto, imposible de diferenciar.
La facilidad con la cual se forma este enlace covalente es explicada de manera sencilla por la capacidad del grupo central para deformar la nube de electrones del ligando, esta capacidad es tanto mayor cuanto mayor es la relación carga/radio del mismo.
Esto permite deducir por qué los cationes, y en especial aquellos con mayor carga y menor tamaño son los que forman complejos con mayor facilidad.

Esta última regla tiene, sin embargo, algunas desviaciones: por ejemplo el cromo Cr3+
que tiene un radio iónico de 0.62 Å forma complejos con mayor facilidad que el aluminio Al3+
, que posee un radio iónico de 0.45 Å. En estos casos cabe considerar el impedimento que poseen determinados aniones o moléculas de gran tamaño para acercarse lo suficiente como para formar el enlace con los orbitales vacíos del grupo central.
La posibilidad de que se forme un enlace entre ligando y grupo central se encuentra condicionada en primer lugar por la existencia o no de orbitales vacíos en el grupo central, y en segundo lugar por la interferencia espacial que se ocasionan entre sí las mismas moléculas de ligando al tratar de acceder a las posiciones donde sería posible que se forme el enlace; esto se conoce como impedimento estérico. Uno se puede hacer una idea bastante buena del impedimento estérico al imaginar un montón de cerditos tratando de acceder a un comedero demasiado pequeño.
Es en gran parte por esto que la química de coordinación está dominada por los metales de transición y de transición interna, ya que se trata de átomos capaces de adquirir elevadas relaciones carga/radio, que poseen en general un gran número de orbitales de valencia desocupados (orbitales d en metales de transición y orbitales f en los de transición interna), y que aun así poseen un radio lo suficientemente elevado como para permitir el acercamiento de un gran número de ligandos.
Ligandos
[editar]Los aniones o moléculas capaces de actuar como ligandos deben poseer átomos que cuenten al menos con un par de electrones de valencia no compartidos. Estos átomos se encuentran en la esquina superior derecha de la tabla periódica, y entre ellos los más importantes son el oxígeno y el nitrógeno, dando paso luego al carbono, fósforo, azufre, cloro, flúor, etc.
Las moléculas que poseen un único átomo donador de electrones se denominan ligandos monodentados, mientras que las que poseen más de un átomo donador reciben el nombre de ligandos polidentados o agentes quelantes. Existe también un tercer tipo de ligandos conocidos genéricamente como ligandos ambidentados que son en realidad ligandos que actúan como monodentados, pero de dos maneras diferentes.
Ligandos monodentados
[editar]Los ligandos de este tipo poseen un único punto de anclaje al núcleo de coordinación, de allí el nombre monodentado que quiere decir un único diente. Comúnmente se trata de moléculas pequeñas, que poseen un único átomo donador de electrones tales como el amoníaco (NH
3), el agua (H
2O), o los aniones halogenuro (X−
), alcóxido (RO−
), o alquilo (R−
) entre otros.
Cuando se forma un complejo de un catión metálico a partir de unión con ligandos monodentados se alteran notoriamente las propiedades de solubilidad del catión, en general esto debido a que el complejamiento provoca un aumento en el tamaño del ion, lo que a su vez se traduce en una disminución en la fuerza de atracción entre el catión y sus contraiones. Esto por lo general provoca un aumento en la solubilidad del ion, o, mejor expresado, una disminución de su tendencia a precipitar.

Ligandos polidentados o agentes quelantes
[editar]Los ligandos de este tipo son capaces de establecer dos o más uniones simultáneas con el núcleo de coordinación, pueden ser bidentados, tridentados, tetradentados etc. A este tipo de ligandos se les suele llamar también "agentes quelantes" un nombre derivado de la palabra griega kela que significa "pinza" porque el tipo de estructura espacial que se forma se asemeja a un cangrejo con el núcleo de coordinación atrapado entre sus pinzas. Muchas veces se utiliza a los agentes quelantes como agentes precipitantes, ya que al ser capaces de establecer dos o más uniones simultáneas también pueden funcionar como "puentes" entre dos o más núcleos de coordinación, facilitando la formación de enormes agregados macromoleculares que precipitan con facilidad.
Entre este tipo de compuestos encontramos por ejemplo a los aniones fosfato (PO3−
4), carbonato (CO2−
3), oxalato (-OOC-COO-), etilendiamina (NH
2-CH
2-CH
2-NH
2) y bipiridina. Un ligando polidentado de enorme importancia por la cantidad de aplicaciones que tiene es el EDTA, el EDTA posee seis sitios de unión.
Ligandos ambidentados
[editar]Este tipo de ligandos podría considerarse un caso especial de los ligandos polidentados, porque poseen más de un átomo capaz de donar pares de electrones no compartidos, sin embargo poseen un tamaño demasiado pequeño como para ser capaces de donar electrones con ambos átomos a la vez, y en lugar de ello se enlazan de una manera ú otra dependiendo de las circunstancias.
Dentro de este grupo encontramos por ejemplo a los aniones tiocianato (S=C=N-), nitrito (O=N-O-) e isotiocianato (NC-S-)
Carga y número de coordinación
[editar]La carga total de un ion complejo se determina por la sumatoria de las cargas del núcleo de coordinación, más la de los ligantes que participan; por ejemplo en el ion hexacianoferrato (III) [Fe(CN)
6]3-, la carga del catión es +3, y cada uno de los iones cianuro posee carga -1, luego:

que es la carga total del ion.
Los ligandos se unen al núcleo de coordinación en una región bastante próxima al mismo llamada esfera de coordinación que es el lugar en el espacio donde es posible que los electrones del ligando interactúen con los orbitales vacíos del grupo central.
Cada uno de los átomos del ligando que accede a la esfera de coordinación para aportar un par de electrones no compartidos se denomina átomo donador.
El número de coordinación de un núcleo de coordinación en es directamente el número de pares de electrones que recibe de los átomos del o de los ligandos. Este valor depende del tamaño del núcleo de coordinación y del tamaño de los ligantes que participan en el complejo. Por ejemplo el hierro Fe3+
se coordina con hasta 6 aniones fluoruro para formar el complejo [Fe(F)
6]3−
(número de coordinación = 6), pero solo puede coordinarse con hasta 4 iones cloruro [Fe(Cl)
4]−
(número de coordinación = 4) debido al tamaño mayor de este último.
Formulación y nomenclatura
[editar]Reglas de formulación
[editar]Para expresar la fórmula de los compuestos de coordinación es conveniente tener presentes las reglas de formulación recomendadas por IUPAC, estas reglas son:
- Los complejos se escriben entre corchetes
- Dentro de los corchetes se escriben primero los cationes, luego los aniones y por último las especies neutras.
- De haber dos o más especies con el mismo tipo de carga, se ordenan alfabéticamente de acuerdo al átomo que se encuentra unido al átomo central.
- Por último y por fuera de los corchetes, se escribe como superíndice la carga total del complejo. Así por ejemplo el hipotético complejo formado por 1Co3+
, 3NH
3 1H
2O, 1Cl−
1F−
se escribiría correctamente como: [CoClF(NH
3)
3(H
2O)]+
Reglas de nomenclatura
[editar]En cuanto a la nomenclatura IUPAC recomienda:
- Tener presente en primer lugar si se trata de un complejo aniónico (con carga negativa) catiónico (con carga positiva) o si se trata de una especie neutra. Por ejemplo:
- [CrCl(NH
3)
5]2+
es un complejo catiónico. - [Co(CN)
6]4−
es un complejo aniónico. - [CuBr
2(NH
3)
2] es un complejo neutro.
- [CrCl(NH
- Al nombrarlo se citan primero los ligandos, y estos en orden alfabético.
- Los ligandos aniónicos se citan por su nombre habitual, por ejemplo H- (hidruro) o ClO−
4 (perclorato). Aunque existen un cierto número de ligandos con nombres especiales, mientras que para los ligandos neutros se utiliza su nombre habitual, con dos excepciones:
| Ligando | Nombre | Tipo |
| F− |
fluoro | Aniónico |
| Cl− |
cloro | Aniónico |
| Br− |
bromo | Aniónico |
| I− |
yodo | Aniónico |
| O− 2 |
oxo | Aniónico |
| OH− |
hidroxo | Aniónico |
| O2− 2 |
peroxo | Aniónico |
| HS− |
mercapto | Aniónico |
| S− 2 |
tio | Aniónico |
| H 2O |
Aqua | Neutro |
| NH 3 |
Ammin | Neutro |
| NO | Nitrosilo | Neutro |
| CO | Cabonilo | Neutro |
- El orden alfabético no considera los prefijos numéricos que indican la presencia de varias moléculas de un mismo ligando. Por ejemplo aqua, diaqua y triaqua van antes que ciano.
- Se utilizan los prefijos di-, tri-, etc., para especificar el número de cada clase de ligando sencillo (unidentado).
- Para ligandos complicados (agentes quelantes polidentados), se usan otros prefijos:
| Cantidad de ligandos | Prefijo |
| 2 | bis |
| 3 | tris |
| 4 | tetraquis |
| 5 | pentaquis |
| 6 | hexaquis |
- El nombre de los ligandos complicados se escribe encerrado entre paréntesis.
- Los ligandos ambidentados reciben un nombre diferente de acuerdo a cuál sea el átomo que se une al grupo central. Por ejemplo:
- NO
2 si se une a través del oxígeno (<:O-NO) se denomina nitrito, pero si lo hace a través del nitrógeno (<:NO
2) se denomina nitro. - SCN si se une a través del azufre (-S-CN) se denomina tiocianato, pero si se une a través del nitrógeno (-NCS) se denomina isotiocianato.
- NO
- Una vez que ya se han nombrado todos los ligandos, se nombra al átomo central de la siguiente manera:
- Si se trata de un complejo ANIONICO se utiliza la raíz del nombre del átomo central seguida de la terminación ATO, y al final entre paréntesis se escribe el estado de oxidación del átomo central con números romanos (Sistema de Stock). Por ejemplo:
- [Fe(CN)
5(H
2O)]2−
: ion aquapentacianoferrato (III)
- [Fe(CN)
- Si se trata de un complejo CATIONICO o NEUTRO no se añade ningún sufijo al nombre del átomo central. Por ejemplo:
- [Ni(CO)
4] tetracarbonilníquel (0) - [Fe(H
2O)
6]2+
ion hexaaquahierro (II)
- [Ni(CO)
- Las sales de iones complejos se denominan como cualquier otra sal, teniendo en cuenta el nombre del anión o catión complejo. Por ejemplo:
- K
4[Fe(CN)
6] hexacianoferrato (II) de potasio - Mg
2[Ni(NCS)
6] hexakis (isotiocianato) niquelato(II) de magnesio - [Co(H
2O)
6]Cl
2 cloruro de hexaaquacobalto(II) - [Cu(NH
3)
4]SO
4 sulfato de tetrammincobre(II) - [CoBr
2(en)
2]Cl cloruro de dibromobis (etilendiamina) cobalto(III) - [Pt(NH
3)
4][PtCl
6] hexacloroplatinato(IV) de tetramminplatino(II)
- K
Complejos metálicos
[editar]Prácticamente todos los compuestos metálicos están formados por algún tipo de compuesto de coordinación (con excepción de los metales en estado de vapor, plasmas y aleaciones). Por lo que el estudio de la química de coordinación es en gran medida equivalente al estudio de la química inorgánica, desde el momento que la química de coordinación es la química de la mayor parte de la tabla periódica. Los átomos e iones metálicos solo existen en la fase condensada rodeados por ligandos.
Las áreas de la química de coordinación de metales pueden ser clasificadas de acuerdo a la naturaleza de sus ligandos. A grandes rasgos estas divisiones son:
- Química de coordinación clásica (o de los "complejos de Werner"): aquí los ligandos se unen a los metales, casi exclusivamente, por medio de pares solitarios de electrones que provienen del grupo principal de átomos del ligando. Ejemplos: H2O, NH3, Cl-, CN-, en-, [Co(EDTA)]−, [Co(NH3)6]Cl3, Fe(C2O4)3]K3
- Química organometálica: los ligandos son compuestos orgánicos sencillos (alquenos, alquinos, alquilos) como así también ligandos de estructura similar a los orgánicos tales como fosfinas, hidruro y carbonilo. Ejemplo: (C5H5)Fe(CO)2CH3
- Química bioinorgánica: los ligandos son compuestos orgánicos producidos por seres vivos, en especial cadenas laterales de aminoácidos, y muchos cofactores tales como las porfirinas. Ejemplos: Hemoglobina, Vitamina B12, Clorofila
- Química de clústeres (grupos), en ésta los ligandos son todos los citados anteriormente y además incluye a otros metales como ligandos. [Ru3(CO)12]
Aunque en muchos casos es difícil clasificar a un caso dentro de un grupo particular y es más fácil interpretarlo como una combinación de varios de ellos. Ejemplos: [Fe4S4(Cisteinil)4]2−, que es en realidad un clúster contenido dentro de una proteína biológicamente activa.
La mineralogía, la tecnología de materiales, y la química del estado sólido (mientras se aplique a iones metálicos); pueden ser consideradas subdivisiones de la química de coordinación, en el sentido de considerar a metales rodeados de ligandos. En muchos casos estos ligandos son óxidos o sulfuros. Es verdad que el foco de la mineralogía, la tecnología de materiales y la química del estado sólido difiere del foco usual de la química de coordinación. Las primeras se ocupan principalmente de estructuras poliméricas, y de las propiedades que se derivan de los efectos colectivos de un enorme número de metales interconectados. Mientras que la segunda, en contraste, se enfoca en la reactividad y propiedades de complejos que contienen átomos metálicos individuales, o pequeños agrupamientos de átomos; pero aun así los metales se encuentran coordinados, y los lineamientos y principios considerados para complejos también se les aplica.
Estructura espacial de los complejos
[editar]Las estructuras moléculares en la química de coordinación se encuentran descritas principalmente por el número de coordinación, es decir por el número de ligandos unidos al grupo central (más específicamente, al número de enlaces sigma entre ligandos y grupo central). Normalmente es posible contar los ligandos unidos, pero algunas veces la cuenta de ligandos puede tornarse un poco ambigua. El número de coordinación se encuentra normalmente comprendido entre uno y nueve, pero no son extraños números de coordinación aun mayores para los lantánidos y actínidos. El número de coordinación va a depender del tamaño, carga, y configuración electrónica del grupo central y de los ligandos.
Los iones metálicos pueden presentar más de un número de coordinación.
La química de los complejos se encuentra dominada por las interacciones entre los orbitales moleculares s y p del ligando y los orbitales d de un ion metálico central. En conjunto los orbitales s, p y d del ion central pueden acomodar 18 electrones (ver la regla de 18 electrones), aunque para elementos del bloque f, esta regla se extiende hasta 32 electrones. El número máximo de coordinación para determinado elemento se encuentra por lo tanto relacionado con su configuración electrónica, (más específicamente con el número de orbitales vacíos que posee), y a la relación entre el tamaño de los ligandos y del grupo central. Grupos centrales grandes y ligandos pequeños permiten números de coordinación elevados, por ejemplo el [Mo(CN)8]4-. Grupos centrales pequeños y ligandos de gran tamaño tienden a desarrollar números de coordinación pequeños, por ejemplo Pt[P(CMe3]2. Es debido precisamente a su gran tamaño, que los lantánidos, actínidos y primeros elementos de transición tienden a desarrollar números de coordinación elevados.
De los diferentes números de coordinación resultan diferentes arreglos estructurales. La mayoría de las estructuras siguen un patrón cuasiesférico, (o, visto de otro modo, como si el grupo central se encontrara en medio de un poliedro y los grupos ligandos se ubicaran en los vértices del mismo). Es en estos puntos donde es posible que se produzca el solapamiento entre los orbitales de los ligandos y el grupo central. Las repulsiones ligando-ligando tienden a dirigir esta organización hacia determinadas geometrías regulares que minimizan las interferencias. Hay sin embargo, numerosos casos de desviaciones de estas organizaciones regulares, por ejemplo en los casos donde se unen ligandos de diferentes tipos, lo que causa diferentes longitudes de enlace, apartando a los ligandos de su organización cuasiesférica, o cuando se producen distorsiones por efectos electrónicos, por ejemplo en la distorsión de Jahn-Teller.
Geometría
[editar]Para los números de coordinación entre dos y nueve los arreglos geométricos más comunes que se presentan en complejos son aquellos que tienden a minimizar las fuerzas de repulsión entre orbitales de la capa de valencia
Se deben notar sin embargo algunas excepciones y previsiones:
- La descripción idealizada para números de coordinación 5, 7, 8 y 9, a menudo es geométricamente indistinta de estructuras alternativas con ángulos L-G-L (ligando-Grupo central-Ligando) ligeramente distintos. El ejemplo clásico de esto es la diferencia entre las estructuras piramidal cuadrada y bipiramidal trigonal.
- Debido a algunos efectos electrónicos especiales, tales como la estabilización de segundo orden Jahn-Teller, ciertas geometrías se ven favorecidas frente a otras estructuras posibles. Por ejemplo, para algunos compuestos con número de coordinación seis, la geometría prismática trigonal se ve favorecida por estabilización y es la adoptada por el complejo en lugar de la octaédrica.
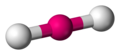
Lineal
[editar]La Lineal es la estructura de menor energía para un número de coordinación dos. En esta disposición el grupo central se encuentra entre los dos grupos ligandos y los tres forman una línea con un ángulo de enlace L-G-L de 180°
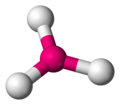
Trigonal plana
[editar]La geometría molecular trigonal plana es la estructura que minimiza las interacciones para un número de coordinación tres. En esta disposición el grupo central se encuentra en el centro de un triángulo equilátero y los grupos ligandos se ubican en los vértices del mismo, con un ángulo de enlace L-G-L de 120°.
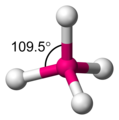
Tetraédrica
[editar]La estructura tetraédrica es la de menor energía posible para un número de coordinación cuatro. En esta disposición el grupo central se encuentra en medio de un tetraedro regular y los grupos ligandos se ubican en los vértices del mismo con un ángulo de enlace L-G-L de 109,5°.
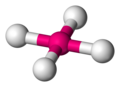
Plano cuadrada
[editar]La geometría molecular plano cuadrada es otra estructura posible para un número de coordinación cuatro, en esta los cuatro ligandos se disponen en un mismo plano en los vértices de un cuadrado. Aparentemente es de energía mayor que la tetraédrica ya que los ángulos L-G-L son de 90°, pero aquí participan repulsiones debidas a orbitales con pares solitarios que se encuentran en las posiciones polares (los pares están en un plano perpendicular al plano que comparten las moléculas en disposición cuadrada). Los metales con configuración electrónica nd8 tienden a adoptar la geometría cuadrada plana.
Bipiramidal trigonal
[editar]La geometría molecular bipiramidal trigonal es la que maximiza los ángulos de separación, y por lo tanto minimiza la energía para un número de coordinación de cinco. se puede ver como dos tetraedros unidos por la base y está muy próxima en energía a su isocoordinada. Esta disposición es anisotrópica, los ligandos en posición ecuatorial se encuentran separados 120° entre sí, pero un ligando ecuatorial se encuentra separado 90° de uno polar.
Piramidal cuadrado
[editar]La geometría molecular piramidal cuadrada se obtiene desplazando ligeramente uno de los vértices polares de una bipirámide trigonal hasta dejarlo en el mismo plano que el formado por dos de los vértices ecuatoriales y el restante vértice polar.
Octaédrica
[editar]La octaédrica es la más típica disposición geométrica para los elementos de transición, y no resulta difícil ver porqué, si pusiéramos una esfera en el interior de un cubo (esfera inscrita), la esfera tocaría las caras del cubo en los vértices de un octaedro. Esta disposición consta de cuatro ligandos colocados en un mismo plano (llamado plano ecuatorial) y un ligando a cada uno de los lados de ese plano en "posición polar", en esta estructura el mínimo ángulo entre ligandos es de 90°.
Prismática trigonal
[editar]La geometría molecular prismática trigonal es la siguiente en estabilidad para un número de coordinación seis, suele ser de menor estabilidad porque implica que los ligandos de los vértices del prisma queden enfrentados unos a otros, esta interferencia se minimiza en la disposición octaédrica (que en cierta forma podría ser considerada un antiprisma trigonal, donde se ha girado la cara superior para que los vértices no queden enfrentados). Por lo general esta estructura se presenta por una estabilización debida a algún otro factor no exclusivamente geométrico, por ejemplo por distorsión forzada de orbitales.
Bipiramidal pentagonal
[editar]La configuración bipiramidal pentagonal es la preferida para un número de coordinación siete, como su nombre lo indica se puede ver como dos pirámides de base pentagonal unidas por la base.
Antiprismática cuadrada
[editar]
La geometría molecular antiprismática cuadrada es la configuración de menor energía entre las tres posibles configuraciones para un número de coordinación ocho, se puede pensar como un cubo en el que se ha girado la cara superior para que los vértices no queden enfrentados.
Bipiramidal hexagonal
[editar]La estructura molecular bipiramidal hexagonal es la siguiente en estabilidad para un número de coordinación ocho.
Tetraédrica triapicada
[editar]
La estructura molecular tetraédrica triapicada es una estructura muy extraña entre los metales de transición, pero para elementos de transición interna resulta ser la estructura que minimiza todas las interacciones entre ligandos y las distorsiones orbitales por lo que se presenta incluso en compuestos muy sencillos tales como el ThCl4. Puede ser racionalizado como un dodecaedro de caras triangulares.
Cúbica
[editar]Es la geométricamente menos estable de las configuraciones para el número de coordinación ocho y prácticamente no existe para los elementos de transición, aunque parece ser bastante común entre los elementos de transición interna, principalmente debido a que los orbitales f(xyz) apuntan hacia los vértices de un cubo, lo que disminuye la distorsión de estos orbitales al interactuar con los ligandos.
Prismática trigonal triapicada
[editar]La Prismática trigonal triapicada es la geometría más regular y estable que existe para un número de coordinación nueve. La aproximación más sencilla para comprender esta estructura tridimensional es imaginarse un prisma trigonal y a media altura insertar un triángulo de modo que los vértices de este queden apuntando al centro de las caras cuadradas del prisma.
Índices de coordinación superiores a 9
[editar]Aunque más de las dos terceras partes de los complejos conocidos presentan índices de coordinación entre 6 y 9, se conocen algunos ejemplos de lantánidos y actínidos con índices de coordinación superiores: 10, 11 o 12.
El número de coordinación 12 presenta un mayor número de ejemplos que el número de coordinación 10 y son muy pocos los descritos para número de coordinación 11. Sin embargo debe mencionarse que, en todos los casos, son complejos que implican ligandos quelato o macrociclo, no se conocen ejemplos de complejos con índices de coordinación mayores que 9 formados por ligandos monodentados.
Para un número de coordinación 12, cabría esperar una estructura regular con forma icosaédrica. Sin embargo, esta estructura prácticamente no existe en la química práctica. Por el contrario, la gran mayoría de las estructuras descriptas corresponden a tetraedros truncados, cubooctaedros o cubos tetraapicados.
Existen trabajos de química computacional que concluyen que en realidad la mejor manera de plasmar los índices de coordinación superiores a 9 para estas estructuras es mediante esquemas de enlace más simples (tetraedro, bipirámide trigonal u octaedro) y que las estructuras pseudoregulares observadas son en realidad una consecuencia de la interacción de los orbitales del ligando quelato con la gran superficie de los orbitales 4f, 5f, 5d o 6d del grupo central, y no solo es debida a la coordinación con los átomos dadores.
Galería de geometrías
[editar]-
 Arreglo espacial en un complejo lineal.
Arreglo espacial en un complejo lineal. -
 Arreglo espacial en un complejo trigonal
Arreglo espacial en un complejo trigonal -
 Arreglo espacial de un complejo tetraédrico.
Arreglo espacial de un complejo tetraédrico. -
 Arreglo espacial en un complejo cuadrado plano
Arreglo espacial en un complejo cuadrado plano -
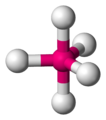 Disposición espacial en un complejo bipiramidal trigonal
Disposición espacial en un complejo bipiramidal trigonal -
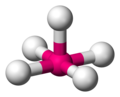 Disposición espacial en un complejo piramidal cuadrado
Disposición espacial en un complejo piramidal cuadrado -
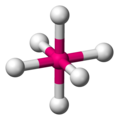 Disposición espacial en un complejo octaédrico
Disposición espacial en un complejo octaédrico -
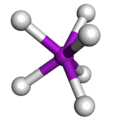 Disposición espacial en un complejo prismático trigonal.
Disposición espacial en un complejo prismático trigonal. -
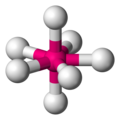 Disposición espacial en un complejo bipiramidal pentagonal
Disposición espacial en un complejo bipiramidal pentagonal -
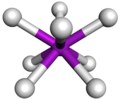 Disposición espacial en un complejo antiprismático cuadrado
Disposición espacial en un complejo antiprismático cuadrado -
![El complejo [UO2(Acetato O, O')3] presenta una geometría bipiramidal hexagonal.](/upwiki/wikipedia/commons/thumb/e/e1/BipirHexg-%28%28UO2%28acetato-O%2CO%27%293%29%29.png/120px-BipirHexg-%28%28UO2%28acetato-O%2CO%27%293%29%29.png) El complejo [UO2(Acetato O, O')3] presenta una geometría bipiramidal hexagonal.
El complejo [UO2(Acetato O, O')3] presenta una geometría bipiramidal hexagonal. -
![El complejo [Y(OH)8]3+ presenta una geometría tetraédrica triapicada.](/upwiki/wikipedia/commons/thumb/a/ac/2Deca%C2%B7angular%28%28Y%28OH%298%29%293%2B.png/120px-2Deca%C2%B7angular%28%28Y%28OH%298%29%293%2B.png) El complejo [Y(OH)8]3+ presenta una geometría tetraédrica triapicada.
El complejo [Y(OH)8]3+ presenta una geometría tetraédrica triapicada. -
![El complejo [U(CNS)8]4- presenta una clara geometría cúbica.](/upwiki/wikipedia/commons/thumb/1/12/Cubica%28%28U%28NCS%298%29%294-.png/120px-Cubica%28%28U%28NCS%298%29%294-.png) El complejo [U(CNS)8]4- presenta una clara geometría cúbica.
El complejo [U(CNS)8]4- presenta una clara geometría cúbica. -
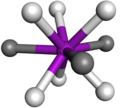 ejemplo de geometría prismática trigonal triapicada, los ligandos que apuntan a las caras del prisma aparecen en color oscuro.
ejemplo de geometría prismática trigonal triapicada, los ligandos que apuntan a las caras del prisma aparecen en color oscuro.










![El complejo [UO2(Acetato O, O')3] presenta una geometría bipiramidal hexagonal.](/eswiki/wiki/Archivo:BipirHexg-((UO2(acetato-O,O%27)3)).png)
![El complejo [Y(OH)8]3+ presenta una geometría tetraédrica triapicada.](/eswiki/wiki/Archivo:2Deca%C2%B7angular((Y(OH)8))3%2B.png)
![El complejo [U(CNS)8]4- presenta una clara geometría cúbica.](/eswiki/wiki/Archivo:Cubica((U(NCS)8))4-.png)

Isomería
[editar]Isomería es la propiedad que relaciona dos o más compuestos que poseen el mismo tipo y número de átomos, pero organizados estructuralmente de manera diferente.
Por razones de claridad se lo ha ubicado en una sección diferente, aunque técnicamente se encuentra dentro del estudio de la geometría de complejos.
La organización estructural de un determinado complejo es, en general, fija y estable, y se encuentra determinada por la disposición de menor energía posible, (esto es más o menos equivalente a decir que adopta la disposición con menores tensiones internas), sin embargo en algunos de estos casos existe más de una arquitectura con energías equivalentes o muy similares, lo que permite que los componentes de ese complejo adopten más de una disposición estructural. En esos casos se presenta la isomería de complejos.
Existe una gran variedad de tipos de isomería en los complejos de coordinación, solo comparable en complejidad por la isomería de los compuestos de carbono.
Estereoisomería
[editar]Estereoisomería es el tipo de isomería que se produce cuando en dos compuestos existen no solo el mismo tipo y número de átomos; sino también el mismo tipo y número de enlaces, pero organizados espacialmente de manera diferente.
El estereoisomerismo puede ser clasificado en:
Isomería geométrica
[editar]La isomería geométrica ocurre en complejos octaédricos y cuadrados planos (no así en los tetraédricos). Cuando dos ligandos ocupan posiciones relativas diferentes uno con respecto a otro. Así cuando dos ligandos se encuentran opuestos uno al otro se dice que son trans y cuando son mutuamente adyacentes se dice que son cis. Cuando tres ligandos idénticos ocupan una de las caras de una disposición octaédrica, se dice que se trata de un isómero facial o fac. Si los tres ligandos se encuentran en el mismo plano que el grupo central, se dice que el isómero es meridional o mer.
Por ejemplo, en un compuesto octaédrico con tres ligandos de un tipo y tres ligandos de otro, existen dos isómeros geométricos: el mer en el cual cada uno de los dos grupos de tres ligandos se encuentra en uno de los meridianos, y el fac en el cual cada grupo de tres ligandos se encuentra en una de las caras del octaédro.
-
![cis-[CoCl2(NH3)4]+](/upwiki/wikipedia/commons/thumb/b/be/Cis-dichlorotetraamminecobalt%28III%29.png/120px-Cis-dichlorotetraamminecobalt%28III%29.png) cis-[CoCl2(NH3)4]+
cis-[CoCl2(NH3)4]+ -
![trans-[CoCl2(NH3)4]+](/upwiki/wikipedia/commons/thumb/5/56/Trans-dichlorotetraamminecobalt%28III%29.png/120px-Trans-dichlorotetraamminecobalt%28III%29.png) trans-[CoCl2(NH3)4]+
trans-[CoCl2(NH3)4]+ -
![fac-[CoCl3(NH3)3]](/upwiki/wikipedia/commons/thumb/6/69/Fac-trichlorotriamminecobalt%28III%29.png/109px-Fac-trichlorotriamminecobalt%28III%29.png) fac-[CoCl3(NH3)3]
fac-[CoCl3(NH3)3] -
![mer-[CoCl3(NH3)3]](/upwiki/wikipedia/commons/thumb/5/54/Mer-trichlorotriamminecobalt%28III%29.png/120px-Mer-trichlorotriamminecobalt%28III%29.png) mer-[CoCl3(NH3)3]
mer-[CoCl3(NH3)3]
![cis-[CoCl2(NH3)4]+](/eswiki/wiki/Archivo:Cis-dichlorotetraamminecobalt(III).png)
![trans-[CoCl2(NH3)4]+](/eswiki/wiki/Archivo:Trans-dichlorotetraamminecobalt(III).png)
![fac-[CoCl3(NH3)3]](/eswiki/wiki/Archivo:Fac-trichlorotriamminecobalt(III).png)
![mer-[CoCl3(NH3)3]](/eswiki/wiki/Archivo:Mer-trichlorotriamminecobalt(III).png)
Isomería óptica
[editar]La isomería óptica se da cuando la imagen especular de un compuesto no es superponible con el compuesto original. Se denomina isomería óptica debido a que los compuestos son ópticamente activos, esto es, que hacen girar el plano de vibración de la luz polarizada. Este comportamiento desigual se produce porque los enlaces, que no son más que grupos de electrones, resuenan de manera diferente sometidos al campo electromagnético que es la luz. En los compuestos ópticamente activos la suma de todas las resonancias da como resultados vectores diferentes, porque los enlaces tienen ciertamente orientaciones diferentes.
se utiliza el símbolo Λ (lambda) para describir la hélice con giro a la izquierda formada por tres ligandos bidentados, tal como se muestra. De manera similar se utiliza el símbolo Δ (delta) como prefijo para describir la hélice con giro hacia la derecha.
-
![Λ-[Fe(ox)3]3−](/upwiki/wikipedia/commons/thumb/d/df/Delta-tris%28oxalato%29ferrate%28III%29-3D-balls.png/110px-Delta-tris%28oxalato%29ferrate%28III%29-3D-balls.png) Λ-[Fe(ox)3]3−
Λ-[Fe(ox)3]3− -
![Δ-[Fe(ox)3]3−](/upwiki/wikipedia/commons/thumb/6/6e/Lambda-tris%28oxalato%29ferrate%28III%29-3D-balls.png/111px-Lambda-tris%28oxalato%29ferrate%28III%29-3D-balls.png) Δ-[Fe(ox)3]3−
Δ-[Fe(ox)3]3− -
![Λ-cis-[CoCl2(en)2]+](/upwiki/wikipedia/commons/thumb/1/12/Delta-cis-dichlorobis%28ethylenediamine%29cobalt%28III%29.png/78px-Delta-cis-dichlorobis%28ethylenediamine%29cobalt%28III%29.png) Λ-cis-[CoCl2(en)2]+
Λ-cis-[CoCl2(en)2]+ -
![Δ-cis-[CoCl2(en)2]+](/upwiki/wikipedia/commons/thumb/8/81/Lambda-cis-dichlorobis%28ethylenediamine%29cobalt%28III%29.png/78px-Lambda-cis-dichlorobis%28ethylenediamine%29cobalt%28III%29.png) Δ-cis-[CoCl2(en)2]+
Δ-cis-[CoCl2(en)2]+
![Λ-[Fe(ox)3]3−](/eswiki/wiki/Archivo:Delta-tris(oxalato)ferrate(III)-3D-balls.png)
![Δ-[Fe(ox)3]3−](/eswiki/wiki/Archivo:Lambda-tris(oxalato)ferrate(III)-3D-balls.png)
![Λ-cis-[CoCl2(en)2]+](/eswiki/wiki/Archivo:Delta-cis-dichlorobis(ethylenediamine)cobalt(III).png)
![Δ-cis-[CoCl2(en)2]+](/eswiki/wiki/Archivo:Lambda-cis-dichlorobis(ethylenediamine)cobalt(III).png)
Isomería estructural
[editar]Este tipo de isomería se da cuando el número y tipo de átomos son iguales, pero enlaces son diferentes entre sí. Existen dos grandes tipos de isomería estructural en los compuestos de coordinación, la isomería de enlace que se produce cuando los ligandos que acceden a la esfera de coordinación son los mismos, y la isomería de esfera de coordinación, en la que los ligandos en la esfera de coordinación son diferentes.
Isomería de enlace
[editar]La isomería de enlace se produce cuando un ligando se puede unir de más de una forma al grupo central, un claro ejemplo es lo que ocurre con los ligandos ambidentados, por ejemplo el NO2 es un ligando ambidentado: se puede unir al grupo central por cualquiera de sus oxígenos (que son equivalentes) o por el nitrógeno.
Isomería de esfera de coordinación
[editar]Este tipo de isomería se produce cuando un compuesto de coordinación alterna los grupos ligandos que acceden a su esfera de coordinación por grupos que se encuentran fuera de la misma en el retículo sólido (ver agua de cristalización). Por ejemplo, el CrCl3(H2O)6 existe en tres formas comunes: [Cr(H2O)6]Cl3 (de color violeta), [Cr(H2O)5Cl]Cl2·H2O (de color verde), y [Cr(H2O)4Cl2]Cl·2H2O (también de color verde). En los compuestos segundo y tercero, el agua ha sido desplazada de la esfera de coordinación por iones cloruro y en su lugar pasa a ocupar posiciones en el retículo sólido del cristal.
Estructura electrónica de los complejos
[editar]Prácticamente todas las propiedades que se observan en los complejos son producto de sus estructuras electrónicas, esto es, son funciones de la manera en que los electrones se organizan dentro de la molécula, tratar de entender entonces como es que estos electrones se encuentran organizados es una buena forma de tratar de empezar a entender las propiedades de los complejos.
A partir de los descubrimientos de Werner, comenzaron a aparecer diferentes modelos teóricos que trataron de explicar los extraños comportamientos observados en los complejos.
Hacia 1929 aparece la teoría del campo cristalino (TCC) propuesta por los físicos Hans Bethe y Van Vleck, que permite explicar de manera conceptualmente muy sencilla el color y las propiedades magnéticas de los complejos, aunque no se correlaciona de buena manera con todos los complejos, ni permite explicar la naturaleza de los enlaces.
Hacia 1933 Linus Pauling introduce la teoría del enlace de valencia (TEV), que trata de explicar el porqué de la direccionalidad de los enlaces en los compuestos y comienza a dar una explicación de la naturaleza de los mismos.
La teoría de orbitales moleculares (TOM) es una consecuencia natural de la teoría del enlace de valencia que avanza sobre los aspectos cuánticos del enlace químico, permite una comprensión mucho más profunda de los fenómenos implicados en la formación de un enlace químico y permite explicar los comportamientos de muchas clases de complejos, sin embargo para muchas aplicaciones resulta demasiado complicada.
La teoría del campo de ligandos (TCL) es un modelo combinado que permite una derivación sencilla de la teoría de orbitales moleculares, utilizando para resolver las ecuaciones formales una aplicación de la teoría de grupos. Goza casi de la misma sencillez conceptual que la teoría de campo cristalino y permite explicar de manera razonablemente precisa una gran cantidad de compuestos.
En todas las disciplinas científicas siempre se trata de utilizar el modelo más sencillo que sirva para explicar cada situación en particular, es por ello que, a pesar de que la teoría de orbitales moleculares es la que brinda una simulación más realista, en general para situaciones sencillas se suele utilizar la teoría de campo cristalino como modelo.
Teoría del campo cristalino (TCC)
[editar]
Esta teoría considera solo la geometría de los orbitales d de un catión central y su interacción con unos ligantes considerados como cargas negativas puntuales. Según este modelo los ligandos son atraídos por la carga positiva del metal, pero al aproximarse generan repulsiones sobre los electrones d del catión deformando los orbitales en los que estos se encuentran. Un orbital deformado presenta una mayor energía que uno con su forma "natural" por lo que los electrones tienden a ocupar posiciones en los orbitales "nativos" siempre que resulte posible, esto es siempre que la diferencia de energía entre los orbitales de mayor y los de menor energía no sea menor que la energía de apareamiento debida a la repulsión de los electrones en un mismo orbital.
Un ejemplo sencillo es lo que ocurre para un complejo octaédrico, si se hace la suposición de que los ligandos avanzan sobre los ejes de un sistema cartesiano tridimensional, los orbitales que se van a ver principalmente afectados son los que tienen componentes principales sobre estos ejes. al ver el gráfico de orbitales d se puede notar que estos orbitales son el d z2 y el dx2-y2. Como consecuencia aumentan su energía y se separan del resto de los orbitales d, formando dos subgrupos de orbitales: el grupo de alta energía eg y el grupo de baja energía t2g.
El grado de separación entre orbitales eg y t2g va a depender de la fuerza de los ligantes, es decir del grado en que estos ligantes sean capaces de deformar los orbitales d. La serie espectroquímica es una tabla empírica que ordena los ligandos de acuerdo al grado de separacíon que causan en los orbitales d, de menor a mayor fuerza son:
I− < Br− < S2− < SCN− < Cl− < NO3− < N3- < F− < OH− < C2O42− < H2O < NCS− < CH3CN < py < NH3 < en < 2,2'-bipiridina < phen < NO2− < PPh3 < CN− < CO
Según este modelo las transiciones electrónicas entre estos orbitales d de diferente energía son las responsables de las absorciones de determinadas longitudes de onda que dan color a los complejos (a menor longitud de onda mayor energía y por tanto mayor fuerza del ligando).
Cuando la separación entre orbitales de baja energía y de alta energía es mayor que la energía de apareamiento de los electrones, todos los electrones tienden a ocupar posiciones en los orbitales de baja energía de acuerdo al principio de Aufbau, formando lo que se conoce como un complejo de bajo espín. Por otro lado si la separación entre orbitales es menor que la energía de apareamiento, los electrones tienden a ocupar todos los orbitales (sean de baja o alta energía) antes de empezar a aparearse de acuerdo a la regla de Hund, formando lo que se conoce como un complejo de alto espín.
De acuerdo a este modelo la cantidad de electrones en condiciones de espín apareado o desapareado, son las responsables de las propiedades magnéticas de los complejos.
Teoría del enlace de valencia (TEV)
[editar]La teoría del enlace de valencia es principalmente un avance geométrico sobre una estructura de Lewis. Básicamente se basa en la idea de que varios orbitales atómicos diferentes pueden combinarse de manera lineal (esto es siguiendo las operaciones del álgebra lineal) para formar orbitales híbridos de iguales energías y con una disposición particular en el espacio.
Linus Pauling, utilizando las ideas de un trabajo previo de Walter Heitler y Fritz London , desarrolla entre 1.930 y 1.940 esta nueva teoría sobre el enlace covalente diciendo que se forma por solapamiento o superposición de dos orbitales atómicos, de modo que los electrones compartidos pertenecen a la vez a los orbitales de los dos elementos enlazados. En otras palabras, esta teoría supone que la formación del enlace covalente ocurre porque se produce el solapamiento o traslape de los orbitales atómicos de los átomos participantes, y que este emparejamiento tiene lugar de modo tal que se produzca el apareamiento de electrones con espines electrónicos de signo contrario; dando lugar a una región espacial de densidad electrónica aumentada común a ambos átomos.
En pocas palabras esta teoría establece que un enlace químico se forma cuando se combinan de manera lineal orbitales de diferentes átomos en determinadas regiones del espacio, sin embargo se distingue de la teoría de orbitales moleculares en que la teoría del enlace de valencia considera a los electrones como pertenecientes a cada átomo en particular.
Para su comprensión es necesario tener en cuenta que únicamente interesarán los orbitales más exteriores de la estructura atómica, y que en la forma espacial que adquieren se ven favorecidas aquellas estructuras de hibridación que maximizan las interacciones para formar enlaces, esto es, que aumentan el solapamiento o traslape de orbitales.
La teoría del enlace de valencia forma parte y complementa a la teoría de orbitales moleculares. La teoría de orbitales moleculares puede predecir propiedades magnéticas (diamagnetismo y paramagnetismo) de una forma más directa, aunque la teoría de enlace de valencia en una forma complicada genera los mismos resultados.
Teoría de orbitales moleculares (TOM)
[editar]La Teoría de orbitales moleculares hace uso de una combinación algebraica lineal de funciones de onda de orbitales atómicos para formar orbitales híbridos, en este sentido se parece a la teoría del enlace de valencia, con la diferencia de que la teoría de orbitales moleculares considera que los orbitales de valencia atómicos desaparecen al formarse un compuesto quedando en su lugar orbitales moleculares que cubren enteramente a la molécula. Un orbital molecular no es más que un orbital de Schrödinger que incluye en su planteamiento a más de un núcleo atómico.
La teoría de orbitales moleculares indica que se forman tantos orbitales moleculares como orbitales atómicos de valencia hay disponibles para formarlos. Así, si se trata de un compuesto que posee dos átomos uno con un orbital s y otro con un orbital s y tres orbitales p, la molécula formada poseerá cinco orbitales moleculares.
Estos orbitales moleculares se dividen en enlazantes, no enlazantes y antienlazantes. Los orbitales enlazantes son aquellos en los que los electrones tienden a pasar la mayor parte del tiempo formando una zona con alta densidad de carga entre los núcleos atómicos, esta zona provoca interacciones atractivas núcleo-electrones-núcleo que mantienen unidos a los átomos entre sí. Un orbital no enlazante es muy similar a un orbital atómico (en la combinación lineal posee mayor proporción de orbital atómico que de orbital molecular), y se encuentra ubicado en estratos profundos de la molécula, un electrón en este tipo de orbitales tiende a pasar aproximadamente el mismo tiempo cerca de cada núcleo sin generar una verdadera densidad de carga entre ambos, por lo que las fuerzas atractivas núcleo-electrones-núcleo son aproximadamente de la misma magnitud que las de repulsión entre núcleos. Un orbital antienlazante es aquel en donde los electrones pasan la mayor parte del tiempo alejados de los núcleos, por lo que las repulsiones núcleo-núcleo que se producen son mucho mayores que las atracciones núcleo-electrones-núcleo.
Los electrones provistos por cada átomo participante se distribuyen dentro de los orbitales moleculares según la regla de Hund, si hay muchos electrones en orbitales enlazantes el compuesto tiene una alta tendencia a formarse y resulta muy estable, y si hay igual número de electrones en orbitales enlazantes que en no enlazantes el compuesto no se forma.
La teoría de orbitales moleculares arriba a resultados similares a los de la hibridación de orbitales atómicos descriptos por la teoría del enlace de valencia, para describir la geometría de moléculas y entre ellas la geometría de complejos, además permite predecir de manera muy acertada la reactividad química, las propiedades magnéticas, el color y hasta la conductividad eléctrica de diferentes substancias. Es muy útil para la confección de modelos computacionales de moléculas, en especial de moléculas extremadamente grandes, y resulta muy fácil de entender para un planteamiento con un número pequeño de átomos. En cierta forma se puede considerar a la teoría del enlace de valencia una especie de caso límite de la teoría de orbitales moleculares. Sin embargo sus soluciones resultan demasiado complejas para resolverlas sobre papel a partir de un número relativamente pequeño de átomos. (Como ejemplo, según esta teoría un complejo con un átomo central del bloque d, y cinco ligandos del bloque p, posee 29 orbitales moleculares, y eso considerando ligandos monoatómicos), por lo que muchas veces resulta más sencillo utilizar la TEV.
Teoría del campo de ligantes (TCL)
[editar]La teoría del campo de ligantes que en cierta forma puede considerarse un híbrido entre la teoría de orbitales moleculares y la teoría del campo cristalino; representa en realidad una aplicación basada en la teoría de grupos de la teoría de orbitales moleculares. Permite describir el enlace, la disposición geométrica de orbitales, las características magnéticas y los colores de los compuestos de coordinación sin las limitaciones que presenta la TCC y con menores dificultades que la TEV.
El análisis por TCL es muy similar al que se hace por orbitales moleculares, pero resulta altamente dependiente de la geometría del complejo, ya que solo hace uso de los orbitales que "geométricamente hablando" pueden formar enlaces, para combinarlos en orbitales moleculares. Esto simplifica notoriamente los planteamientos y los resultados y permite arribar a conclusiones válidas sin tener que recurrir al uso de complicados modelos computacionales.
Coloración de los complejos
[editar]Una sustancia posee color cuando absorbe determinadas longitudes de onda electromagnéticas comprendidas dentro del rango visible.
La absorción de determinadas longitudes de onda es debida a la transición entre dos estados energéticos de los electrones que forman los orbitales de un átomo, o los enlaces de una molécula.
Cada tipo de electrón puede absorber solo determinadas cantidades de energía, debido a la naturaleza del orbital atómico o molecular que ocupa.
Como la diferencia de energía entre dos niveles electrónicos es igual a la energía del fotón absorbido, es posible relacionar esta energía con la longitud de onda del fotón según:

o lo que es lo mismo:

Donde:
- es la energía
- es la constante de Planck
- es la frecuencia de la onda
- es la velocidad de la luz
- es la longitud de onda





Luego cada transición electrónica absorbe determinadas longitudes de onda de luz. Si la transición absorbe longitudes de onda dentro del rango visible (420 a 750 nm), entonces el compuesto, al ser iluminado con luz blanca, se ve coloreado; y precisamente del color complementario al color absorbido. Por ejemplo un compuesto que absorbe luz roja mostrará un color compuesto por los colores azul y verde que no son absorbidos, uno que absorba el color verde mostrará color violeta (rojo y azul), uno que absorba azul mostrará color amarillo (verde y rojo), etc.
Los colores notablemente intensos y vistosos de los compuestos de coordinación están determinados por la diferencia de energía (D) entre los conjuntos de orbitales eg y t2g en sus iones complejos. Cuando el ion absorbe luz en el intervalo visible, los electrones son excitados (saltan) del nivel de energía más bajo t2g al más alto eg.
Por ejemplo consideremos el ion [Ti(H2O)6]3+, el cual da lugar a disoluciones púrpuras en agua. El ion hidratado Ti3+ es un ion d1, con el electrón d en uno de los tres orbitales t2g de menor energía. La diferencia de energía (D) entre los orbitales eg y t2g en este ion corresponde a la energía de los fotones que abarca el intervalo verde y amarillo. Cuando la luz blanca incide sobre la disolución, estos colores de la luz se absorben, y el electrón salta a uno de los orbitales eg. Se transmite luz roja, azul y violeta, así que la disolución se ve púrpura.
Los espectros de absorción muestran las longitudes de onda absorbidas por un ion metálico con diferentes ligandos, y por iones metálicos diferentes con el mismo ligando.
A partir de datos como estos, es posible inferir la estructura de los orbitales d que intervienen en el complejo ya que relacionamos la energía de la luz absorbida con los valores de D (diferencia de energía entre orbitales de alto y bajo espín) y surgen dos observaciones importantes:
Para un ligando dado, el color depende del estado de oxidación del ion metálico. Una disolución del ion [V(H2O)6]2+ es violeta, y una disolución del ion [V(H2O)6]3+ es amarilla. Para un ion metálico dado, el color depende del ligando. Esta observación permite clasificar a los ligandos en la serie espectroquímica arriba comentada.
| FeII | FeIII | CoII | CuII | AlIII | CrIII | |
|---|---|---|---|---|---|---|
| Ion hidratado | [Fe(H2O)6]2+ Verde pálido Soluble |
[Fe(H2O)6]3+ Amarillo/Marrón Soluble |
[Co(H2O)6]2+ Rosado Soluble |
[Cu(H2O)6]2+ Azul Soluble |
[Al(H2O)6]3+ Incoloro Soluble |
[Cr(H2O)6]3+ Verde Soluble |
| OH–, diluido | [Fe(H2O)4(OH)2] Verde oscuro Forma precipitado |
[Fe(H2O)3(OH)3] Marrón Forma precipitado |
[Co(H2O)4(OH)2] Azul verdoso Forma precipitado |
[Cu(H2O)4(OH)2] Azul Forma precipitado |
[Al(H2O)3(OH)3] Blanco Forma precipitado |
[Cr(H2O)3(OH)3] Verde Forma precipitado |
| OH–, concentrado | [Fe(H2O)4(OH)2] Verde oscuro Forma precipitado |
[Fe(H2O)3(OH)3] Marrón Forma precipitado |
[Co(H2O)4(OH)2] Azul verdoso Forma precipitado |
[Cu(H2O)4(OH)2] Azul Forma precipitado |
[Al(OH)4]– Incoloro Soluble |
[Cr(OH)6]3– Verde Soluble |
| NH3, diluido | [Fe(H2O)4(OH)2] Verde oscuro Forma precipitado |
[Fe(H2O)3(OH)3] Marrón Forma precipitado |
[Co(H2O)4(OH)2] Azul verdoso Forma precipitado |
[Cu(H2O)4(OH)2] Azul Forma precipitado |
[Al(H2O)3(OH)3] Blanco Forma precipitado |
[Cr(H2O)3(OH)3] Verde Forma precipitado |
| NH3, concentrado | {[Fe(H2O)4(OH)2] Verde oscuro Forma precipitado |
[Fe(H2O)3(OH)3] Marrón Forma precipitado |
[Co(NH3)6]2+ Pajizo Soluble |
[Cu(NH3)4(H2O)2]2+ Azul intenso Soluble |
[Al(H2O)3(OH)3] Blanco Forma precipitado |
[Cr(NH3)6]3+ Verde Soluble |
| CO32– | FeCO3 Verde oscuro Forma precipitado |
[Fe(H2O)3(OH)3] Marrón Forma precipitado Libera burbujas de CO2 |
CoCO3 Rosado Forma precipitado |
CuCO3 Azul verdoso Forma precipitado |
Propiedades magnéticas de los complejos
[editar]En general las propiedades magnéticas dependen del número de electrones desapareados que posea el complejo. Cuando haya uno o más electrones desapareados, el complejo será paramagnético y se verá atraído por los campos magnéticos en grado proporcional al número de electrones desapareados. Si no hay electrones desapareados, el compuesto será diamagnético y se verá ligeramente repelido por los campos magnéticos. Como al estar desapareados, la energía del sistema es menor, si el desdoblamiento energético es pequeño, es más favorable la situación en la cual los electrones están desapareados, ocupando los orbitales d superiores e inferiores (configuración de alto espín) , mientras que si el desdoblamiento es grande, los electrones estarán apareados en los niveles d inferiores (configuración de bajo espín). En el primer caso, el complejo es fuertemente paramagnético, mientras que en el segundo solo lo es débilmente (5 e- desapareados frente a 1).
Propiedades químicas de los complejos
[editar]Los complejos pueden presentar una amplia variedad de propiedades químicas dependiendo del tipo de reacción en la que participen:
- Reacciones de transferencia de electrones
- Los grandes tipos de reacciones más comunes de transferencia de electrones en las cuales participan los complejos son dos: transferencia de electrones dentro de la esfera de coordinación y transferencia de electrones con el exterior de la esfera de coordinación. Dentro de estos tipos de reacciones la transferencia de electrones se produce por mecanismos redox clásicos, o por medio de un puenteo de ligando, en el cual un ligando con dos pares solitarios se encuentra unido a dos centros de coordinación diferentes, los electrones son transferidos desde uno de los centros de coordinación al otro a través del ligando.
- Reacciones de Intercambio de ligando (degenerado)
- Un importante indicador de la reactividad de un complejo es la tasa de intercambio entre ligandos degenerados (iguales). Por ejemplo, la tasa de intercambio entre las moléculas de agua que participan en complejos de tipo [M(H2O)6]n+, varía en más de 20 órdenes de magnitud entre los complejos más estables y los más inestables. Aquellos complejos en los cuales los ligandos son liberados y reenlazados con gran rapidez se clasifican como lábiles. Tales complejos lábiles pueden ser muy inestables termodinámicamente. Un complejo metálico lábil típico posee una baja carga (p.ej. Na+), electrones en orbitales d que son antienlazantes con respecto a los ligandos (Zn2+), o un bajo grado de carácter covalente en el enlace (Ln3+, donde Ln es un lantánido cualquiera). La inestabilidad o labilidad de un complejo metálico también depende del tipo de configuración electrónica que presenta. Por ejemplo los complejos de alto espín de Fe(II) y Co(III) son lábiles, mientras que los complejos de bajo espín análogos son inertes. El Cr (III) puede existir únicamente en un estado de bajo espín (cuarteto), el cual es inerte debido a la ausencia de electrones en orbitales que son antienlazantes en la relación Metal-Ligando, más algo de estabilización debida al campo de ligandos asociado a la configuración d3.
- Procesos asociativos
- Los complejos que poseen orbitales vacíos o semillenos presentan además la capacidad de reaccionar con otros sustratos. La mayor parte de los sustratos poseen un estado basal de tipo singlete; esto es que poseen pares solitarios (por ejemplo agua, aminas, éteres), de modo que tales sustratos requieren de orbitales vacíos para ser capaces de reaccionar con el núcleo de coordinación. Algunos sustratos, como por ejemplo el oxígeno molecular poseen un estado basal de tipo triplete, lo que provoca que los núcleos de coordinación metálicos con orbitales semillenos presenten una fuerte tendencia a reaccionar con tales sustratos. Es posible decir que el oxígeno molecular también presenta pares solitarios, de modo que es capaz de reaccionar como una base de Lewis normal.
Si se elige cuidadosamente los ligandos que rodean al núcleo de coordinación, este grupo central puede ser utilizado para catalizar la transformación de otras moléculas o puede ser utilizado como indicador o sensor.
Véase también
[editar]Bibliografía
[editar]- Brown TL, LeMay E Jr, Bursten BE. (2009)Chemistry: The Central Science (11th Edition). Prentice-Hall. ISBN 0-13-600617-5
- Chang R. (2002) Química - séptima edición. McGraw-Hill Interamericana. ISBN 1941038940
- ¿Puede la teoría de enlace de valencia ayudar a comprender las geometrías de los complejos que poseen elevados índices de coordinación? Terrón A., García-Ruso A, Barceló-Oliver M. An. Quím. 2008, 104(1), pp 42−46.
Referencias
[editar]
| Control de autoridades |
|
|---|
 Datos: Q107434516
Datos: Q107434516