
Fosfonato

Los fosfonatos, ácidos fosfónicos, organofosfitos o simplemente fosfitos son compuestos orgánicos organofosforados que pueden considerarse formalmente como las sales o los ésteres del ácido fosforoso (H3PO3). Son compuestos organofosforados caracterizados por un enlace estable de carbono-fósforo (C-P), que generalmente resiste la descomposición bioquímica, térmica y fotoquímica. Los fosfonatos son compuestos δ4λ5 con estado de oxidación formal +3.

Debido a la tautomería que se da en el ácido fosforoso, podemos tener dos grupos de fosfonatos, los que contienen los grupos R-PO(OH)2 o R-PO(OR')2 (o su versiones "ácidas" con un H en vez de un radical R) o los que contienen el grupo P(OR')3(donde R'= alquilo, arilo...). Tradicionalmente, se han llamado indistintamente fosfitos y fosfonatos a ambos grupos, pero la IUPAC en 2005, decidió llamar al primer tautómero (H2PHO3) ácido fosfónico, y al segundo (H3PO3) ácido fosforoso, por lo que al primer grupo debería denominarse como fosfonatos o ácidos fosfónicos y al segundo como fosfitos o ésteres de fosfito.
Los bifosfonatos fueron sintetizados por primera vez en 1897 por Von Baeyer y Felix Hoffmann. Un ejemplo de bifosfonato es el HEDP. Desde el trabajo de Schwarzenbach en 1949, los ácidos fosfónicos son conocidos como efectivos agentes quelantes. La introducción de un grupo amino en la molécula para obtener aminofosfonatos (-NH2-C-PO(OH)2) aumenta las habilidades para atrapar metales por parte del fosfonato. Algunos ejemplos de tales compuestos son el EDTMP y el DTPMP. Estos fosfonatos comunes son los análogos estructurales de los bien conocidos aminopolicarboxilatos NTA, EDTA, y DTPA. La estabilidad de los complejos de metal se incrementa con el incremento en el número de grupos ácido fosfónico. Los fosfonatos son altamente solubles en agua, mientras que los ácidos fosfónicos son sólo ligeramente solubles. Los fosfonatos no son volátiles y son poco solubles en disolventes orgánicos.[1]
Existen multitud de compuestos comercialmente importantes dentro del grupo de los fosfonatos, destacando el glifosato (la molécula activa del herbicida "Roundup") y el etefón, un regulador del crecimiento vegetal ampliamente utilizado. Algunos bisfosfonatos son medicamentos populares para el tratamiento de la osteoporosis..[2] Además, debido a la versatilidad que da la química de los fosfonatos, se suelen utilizar como reactivos de partida para su posterior oxidación a fosfatos orgánicos, como es el caso de la síntesis del medicamento antiviral que se emplea para el tratamiento de la infección por el VIH, el tenofovir.
Aunque muchos compuestos de fósforo son reconocidos como reactivos importantes en química orgánica, los fosfonatos en sí han sido prácticamente descuidados. El uso más importante del ácido H-fosfónico es la producción de fosfonatos que se utilizan en el tratamiento del agua. El ácido fosforoso y sus sales de amonio, sodio y potasio y sales de mono y di-potasio están clasificadas como bioplaguicidas por la Agencia de Protección Ambiental de EE. UU. y por la UE. Tienen baja toxicidad en el medio ambiente y son más conocidos por su alta eficacia en el control de enfermedades causadas por oomicetos como Phytophthora, Plasmopara y Pythium.[3][4]

Abundancia en la naturaleza
[editar]
El fósforo es un nutriente esencial para todos los organismos vivos, requerido para la síntesis de ácidos nucleicos, fosfolípidos y otros numerosos metabolitos. En la mayoría de los organismos, la fuente preferida de fósforo es el fosfato inorgánico (Pi). Sin embargo, debido a que la mayoría de las sales de fosfato son altamente insolubles, este ion rara vez está disponible en concentraciones que respalden el crecimiento desenfrenado. Por lo tanto, a pesar de que el fósforo es el undécimo elemento más abundante en la corteza terrestre, es un nutriente limitante en la mayoría de ecosistemas.[5] Como resultado, la naturaleza ha desarrollado sistemas de transporte de fosfato altamente eficientes, así como sistemas para la adquisición de fósforo de esencialmente todas las biomoléculas conocidas que contienen este elemento.[6] Sin embargo, estudios recientes sugieren que otros compuestos de fósforo menos estudiados también pueden ser importantes en la biosfera.[7] Entre estos se encuentran los fosfonatos y fosfinatos, compuestos caracterizados por la presencia de enlaces carbono-fósforo (C-P) altamente estables en lugar de los enlaces lábiles de carbono-oxígeno-fósforo que se encuentran en las biomoléculas más conocidas que contienen fósforo.
El aminofosfonato natural ciliatina (nombre original) o ácido 2-aminoetilfosfónico (2-AEP), que es un análogo del aminoácido β-alanina y el aminosulfonato taurina, fue el primer fosfonato identificado (en 1959)[8] y se encontró posteriormente como el grupo principal de fosfonolípidos producidos por muchos otros microorganismos, animales, e incluso plantas, donde está localizado en las membranas.[9] Los fosfonatos son bastante comunes entre diferentes organismos, desde procariotas a eubacterias, fungi, moluscos, insectos y otros.[10]
También están ampliamente distribuidos entre formas de vida más primitivas, incluidos muchos invertebrados marinos, y constituyen un componente significativo del reservorio de fósforo orgánico disuelto en los océanos. Prácticamente todos los compuestos biogénicos C–P se sintetizan por una vía en la que el paso clave es la reorganización intramolecular del fosfoenolpiruvato a fosfonopiruvato. Sin embargo, la escisión del enlace C–P por microorganismos degradantes es catalizada por una serie de enzimas C–P liasa, C–P hidrolasas y otras de mecanismo aún no caracterizado. La expresión de algunas de las vías del catabolismo de fosfonato está controlada por los niveles ambientales de P inorgánico (Pi), pero para otros es independiente del Pi. Los datos indican la más que probable importancia del P en forma de fosfonato en el ciclo global del fósforo biogeoquímico y, por extensión, su papel en la productividad marina y en la dinámica del carbono y el nitrógeno en los océanos.[11] El descubrimiento de que los fosfonatos forman alrededor del 10% del fósforo disuelto y particulado en los océanos, ha demostrado que la disponibilidad de fósforo es un determinante clave de la productividad del fitoplancton marino.[12] Por lo tanto, parecen ser un recurso importante de este elemento para los organismos acuáticos; sin embargo, la comprensión de su utilización por el fitoplancton eucariota es muy limitada. Lo más probable es que ocurran en forma de polisacáridos esterificados con ácido metilfosfónico y ácido 2-hidroxietilfosfónico. Estos compuestos se han encontrado principalmente en Nitrosopumilus maritimus, uno de los organismos más abundantes del planeta y residente de las regiones ricas en oxígeno de los océanos abiertos. Hasta el 4% del metano en la Tierra proviene de las aguas ricas en oxígeno a través de la división del enlace carbono-fósforo altamente no reactivo en el metilfosfonato.[13]
Aunque su síntesis en protozoos se descubrió hace más de 50 años, el alcance y la diversidad de la producción de fosfonato en la naturaleza sigue siendo poco caracterizada. Desde entonces, la fosfonoalanina y el 1-hidroxi-2-aminoetilfosfonato también se han encontrado como el grupo principal de fosfonolípidos.[14] El 2-AEP y el metilfosfonato son componentes de los fosfonoglucanos producidos por arqueas, bacterias, protozoos e invertebrados. Además, tanto el 2-AEP como el 2-hidroxietilfosfonato (2-HEP) también se han encontrado unidos a los azúcares de las proteínas glicosiladas de las eucariotas inferiores.[15] A pesar de su ubicuidad, la función biológica de las macromoléculas de fosfonato sigue siendo un misterio sin resolver. No se han encontrado en la naturaleza bifosfonatos o polifosfonatos.
Además de los fosfonolípidos, se aislaron una gran variedad de fosfonatos naturales de molécula pequeña durante el siglo XX.[7] Por consiguiente, todos los compuestos descritos durante este período de tiempo tienen bioactividades conocidas: Fosfomicina (antibiótico),[16] bialafos (herbicida),[17] fosalacina (herbicida),[18][19] trialafos,[20] fosmidomicina,[21] las plumbemicinas (antibióticos),[22] y fosfonoclorina (antibiótico)[23] se aislaron originalmente mediante ensayos de inhibición del crecimiento bacteriano. La inhibición del crecimiento fúngico también se usó, consiguiendo aislar los antifúngicos fosfazinomicinas[24] y rizocticinas.[25] También se descubrió la fosfonotrixina, para la prevención de la germinación de las semillas.[26]
La biosíntesis de la mayoría de fosfonatos comienza a partir de la reorganización del fosfoenolpiruvato en fosfonopiruvato, una reacción catalizada por la enzima fosfoenolpiruvato mutasa. En este proceso de equilibrio, la termodinámica favorece al fosfoenolpiruvato por un factor de al menos 500. Por lo tanto, el fosfonopiruvato tiene que convertirse rápidamente en compuestos metabólicamente útiles, favoreciendo ese desplazamiento mediante reacciones irreversibles. En consecuencia, es un sustrato clave en la biosíntesis de ácido 2-aminoetilfosfónico, fosfonoalanina, ácido 2-hidroxietilfosfónico, fosfonoacetaldehído, ácido fosfonometilmálico y ácido 2-ceto-4-hidroxi-5-fosfonopentanoico. La mayoría de las enzimas involucradas en la producción de estos compuestos han sido aisladas y caracterizadas y revisadas exhaustivamente.[27]
Propiedades básicas
[editar]Las sales de fosfonato son el resultado de la desprotonación de ácidos fosfónicos, que son ácidos dipróticos:[1][28]
- (M= Na, K; fosfonato monosódico o monopotásico)
- (M= Na, K; fosfonato disódico o dipotásico)


El anión fosfito (HPO3-2) es un ion poliatómico con un átomo de fósforo en estado de oxidación +3. Tiene geometría tetraédrica. Las numerosas sales de fosfito, tales como el fosfito de potasio, son altamente solubles en agua. La unión puede explicarse utilizando estructuras resonantes (hay tres estructuras equivalentes que contienen un doble enlace), deslocalizando efectivamente las cargas negativas entre átomos de oxígeno equivalentes. Cuatro estructuras resonantes de un ion fosfito:[29]

Los ésteres de fosfonato son el resultado de la condensación de ácidos fosfónicos con alcoholes.
Tautomería de los H-fosfonatos
[editar]
Al igual que se ha comentado anteriormente con el ácido fosforoso, los H-fosfonatos, como los H-fosfinatos, y los óxidos de fosfina secundarios, muestran un equilibrio tautomérico prototrópico entre la forma tautomérica del óxido (O=PH<) tetracoordinado y la forma hidróxido (HO-P<) tricoordinada (un átomo de hidrógeno se mueve de un átomo de P al oxígeno). En particular, estas especies tienen una reactividad diferente, que está influenciada por el efecto de los sustituyentes sobre el tautómero dominante en la mezcla de equilibrio. Para comprender el comportamiento de estas reacciones químicas de los compuestos, es clave investigar la interconversión entre los dos tautómeros y el efecto de los medios circundantes en las moléculas para determinar la termodinámica, la cinética y el mecanismo del equilibrio tautomérico. La reactividad de los derivados H-fosfonatos sugiere la presencia de ambas formas en la disolución. Los H-fosfonatos con sustituyentes electrodonadores existen en un equilibrio desplazado hacia la forma fosfonato, mientras que aquellos con sustituyentes electroatractores tienen una fuerte preferencia hacia la forma fosfito. Además, se encontró una relación logarítmica entre esta estabilidad y la permitividad relativa del medio disolvente, siendo la forma H-fosfonato más estable en disolventes que tienen mayor permitividad relativa.[30]
Los diésteres de H-fosfonato, o dialquil H-fosfonatos, como el dimetilfosfito o el dietilfosfito, se presentan únicamente en la forma tautomérica fosfonato, no hay pruebas de su existencia en la forma fosfito.[31]
Síntesis
[editar]A partir de PCl3
[editar]Los fosfitos se preparan típicamente tratando el tricloruro de fósforo con un alcohol. Dependiendo de los detalles sintéticos, esta alcohólisis puede dar a los dialquilfosfitos:[32]

Alternativamente, cuando la alcohólisis se realiza en presencia de aceptores de protones, se obtienen los derivados de trialcoxi de simetría C3:[33]

Por transesterificación
[editar]Los ésteres de fosfito también se pueden preparar por transesterificación, ya que experimentan intercambio de alcohol al calentarse con otros alcoholes.[34] Este proceso es reversible y puede usarse para producir fosfitos de alquilo mixtos. Los fosfonatos (y otros ésteres de fósforo) también pueden ser sintetizados vía una reacción de transesterificación catalizada por catalizadores orgánicos. Se ha reportado que los carbenos N-heterocíclicos catalizan la reacción eficientemente.[35]
A partir de ácido fosfónico
[editar]La mayoría de los procesos comienzan con ácido fosforoso (también conocido como ácido fosfónico, H3PO3), explotando su enlace reactivo P-H.[28][2]
El ácido fosfónico puede ser alquilado en condiciones de Mannich para dar fosfonatos aminometilados, que son útiles como complejos. Un ejemplo es la preparación industrial de ácido aminotris(metilenfosfónico) o ATMP:

El ácido fosfónico también puede ser alquilado con derivados del ácido acrílico para proporcionar ácidos fosfónicos funcionalizados con carboxilo. Esta reacción es una variante de la adición de Michael:

En el acoplamiento de Hirao, los dialquilfosfitos (que también pueden verse como diésteres de ácido fosfónico: (O=P(H)(OR)2) experimentan una reacción de acoplamiento catalizada por paladio con un haluro de arilo para formar un fosfonato.
Por reducción
[editar]El ácido H-fosfónico reacciona con trimetilclorosilano en presencia de trietilamina para dar tris(trimetilsilil)fosfito con un rendimiento del 78-86%:[36]
![{\displaystyle \mathrm {HP(O)(OH)_{2}\ +\ (CH_{3})_{3}SiCl\ {\xrightarrow[{}]{Et_{3}N}}\ P[OSi(CH_{3})_{3}]_{3}} }](https://wikimedia.org/eswiki/api/rest_v1/media/math/render/svg/75eeee899a9bba22926d529b84ecf53a36df375d)
La sililación se produce porque el grupo sililo se transfiere fácilmente al átomo de oxígeno cargado negativamente debido a la fuerte afinidad del silicio por el oxígeno.
Reacción de Michaelis-Arbuzov
[editar]Los ésteres fosfónicos pueden ser sintetizados usando la reacción de Michaelis-Arbuzov. En un estudio, un α-aminofosfonato es preparado por condensación de benzaldehído, anilina, y fosfito de trimetilo, catalizado por triflato de cobre en una síntesis en un solo reactor.[37]
Por ejemplo, el yoduro de metilo cataliza la conversión de trimetilfosfito en el éster de metilfosfonato de dimetilo (DMMP):

Estos ésteres pueden hidrolizarse al ácido (Me = metilo):

En la reacción de Michaelis-Becker, un diéster de hidrógenofosfonato se desprotona primero y el anión resultante es alquilado.
Reacción de Kabachnik-Fields
[editar]La reacción de Kabachnik–Fields es una reacción orgánica que consiste en la formación de un α-aminofosfonato. Los aminofosfonatos son objetivos importantes en síntesis orgánica como análogos de α-aminoácidos bioisostéricos. Esta reacción fue descubierta de manera independiente por Martin Izrailevich Kabachnik[38] y Ellis K. Fields[39] en 1952:
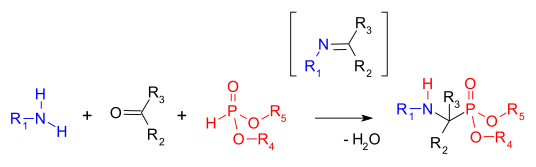
Kabachnik-Fields Reaction

Una vez obtenido el aminodialquilfosfonato, por hidrólisis con un ácido, se puede obtener el aminofosfonato.
Reactividad
[editar]Hidrólisis
[editar]Los ésteres de fosfonato son generalmente susceptibles a la hidrólisis tanto en condiciones ácidas como básicas. La escisión del enlace P-C o P-H no es fácil de lograr.

Reacción de Horner–Wadsworth–Emmons
[editar]En síntesis orgánica, los fosfonatos son usados en la reacción de Horner–Wadsworth–Emmons. En la reacción de Horner-Wadsworth-Emmons, los dialquilfosfonatos se desprotonan para dar carbaniones estabilizados, que reaccionan con aldehídos para dar alquenos-E con la eliminación de un dialquilfosfato:

The Horner–Wadsworth–Emmons reaction

Reacción de Pudovik
[editar]La reacción de Pudovik es un método para preparar α-aminometilfosfonatos. En condiciones básicas, el enlace fósforo-hidrógeno (P-H) de un dialquilfosfito, (RO)2P(O)H, se adiciona al doble enlace carbono-nitrógeno (C=N) de una imina (una reacción de hidrofosfonilación):[40]

Una vez generado el aminofosfito, se pueden eliminar los ésteres por hidrólisis en medio ácido.
Adición a nitrilos
[editar]Los dialquil H-fosfonatos se adicionan a nitrilos en presencia de HCl para dar derivados de aminoalquilbifosfonatos:[31]

Cuando la reacción es llevada a cabo en atmósfera de hidrógeno en presencia de un catalizador de Níquel-Raney, la hidrogenación del nitrilo a azometina tiene lugar en primer lugar, seguida de la adición al dialquil H-fosfonato:[31]

Reacción de Abramov
[editar]La reacción de Abramov se trata de otra reacción de adición de trialquilfosfitos a compuestos de carbonilo, formando α-hidroxifosfonatos. En términos de mecanismo, la reacción implica el ataque del átomo de fósforo nucleófilo sobre el carbono carbonílico:[40]

Reacción de Atherton-Todd
[editar]La reacción de Atherton-Todd es una de las reacciones más características de los diésteres de ácido H-fosfónico. Es una ruta para la oxidación de H-dialquilfosfonatos a los dialquil clorofosfatos altamente reactivos:[31]

La reacción se usa ampliamente in situ en condiciones suaves para la síntesis de una gran cantidad de compuestos biológicamente activos como fosfatos y amidofosfatos.
Subclases estructurales
[editar]En función del número de grupos fosfonato que tenga la molécula, o si se sustituye el átomo de oxígeno por otros heteroátomos, se pueden subclasificar en:
Bifosfonatos
[editar]
Los compuestos que contienen 2 grupos fosfonato en la misma molécula se conocen como bisfosfonatos. Serían el equivalente a un ácido dicarboxílico, en el que se sustituyen los grupos carboxilo por fosfonato. Los bifosfonatos más destacados son geminales. Fueron sintetizados por primera vez en 1897 por Von Baeyer y Hofmann y ahora forman la base de una clase importante de medicamentos, utilizados para tratar la osteoporosis y enfermedades similares. Los ejemplos incluyen al ácido etidrónico (HEDP), que se prepara a partir de ácido fosforoso y anhídrido acético:[2]

Destacan también otros medicamentos para la osteoporosis: ibandronato, risedronato, alendronato, zoledronato...
Polifosfonatos
[editar]Al igual que los ácidos policarboxílicos, cuando hay más de dos grupos fosfonato en la misma molécula se pueden llamar polifosfonatos.
Tiofosfonatos
[editar]

Existen diversos tiofosfonatos y ésteres de tiofosfonato. Este grupo funcional consiste en sustituir un átomo de oxígeno por un azufre. Son un componente reactivo de muchos pesticidas y agentes nerviosos. Los tiofosfonatos sustituidos pueden tener 2 isómeros estructurales principales, ya sea uniéndose el grupo orgánico a través del átomo de O o de S para dar las formas de tiona o tiol respectivamente. Esta es una propiedad que comparten con grupos funcionales relacionados, como los ácidos tiocarboxílicos y los organotiofosfatos.
Fosfonamidatos
[editar]Los fosfonamidatos están relacionados con los fosfonatos mediante la sustitución de un átomo de oxígeno por un nitrógeno. Son un grupo funcional raramente encontrado, el agente nervioso Tabún es un ejemplo.
Compuestos fosfonato
[editar]Cabe destacar los siguientes fosfonatos más característicos:
| Abreviatura | Nombre químico | Número CAS | Estructura química |
|---|---|---|---|
| AMPA | Ácido aminometilfosfónico | 1066-51-9 | 
|
| AEP | Ácido 2-aminoetilfosfónico | 2041-14-7 |
|
| DMMP | Metilfosfonato de dimetilo | 756-79-6 | 
|
| HEDP | Ácido 1-hidroxietiliden-1,1-difosfónico | 2809-21-4 | 
|
| ATMP | Ácido aminotris(metilenfosfónico) | 6419-19-8 | .png)
|
| EDTMP | Ácido etilendiaminotetra(metilenfosfónico) | Sal de sodio: 15142-96-8 |

|
| DTPMP | Ácido dietilentriaminopenta(metilenfosfónico) | Sal de sodio: 22042-96-2 |
.png)
|
Otros fosfonatos conocidos son:
- TDTMP: Ácido tetrametilendiaminotetra(metilenfosfónico).
- HDTMP: Ácido hexametilendiaminotetra(metilenfosfónico).
- PBTC: Ácido fosfobutan-tricarboxílico.
- PMIDA: Ácido N-(fosfometil)iminodiacético.
- CEPA: Ácido 2-carboxietilfosfónico.
- HPAA: Ácido 2-hidroxifosfonocarboxílico.
- AMP: Ácido amino-tris-(metilen-fosfónico).
- Glifosato
- Fosmidomicina
- Foscarnet
Propiedades y usos
[editar]Los fosfonatos son agentes quelantes efectivos que se unen fuertemente a iones metálicos divalentes y trivalentes, evitando que formen precipitados insolubles, y eliminando sus propiedades catalíticas. Son estables bajo condiciones fuertes. Un uso industrial importante de los fosfonatos es en aguas de refrigeración, sistemas de desalinización, y en campos de petróleo para impedir la formación de precipitados. En la manufactura de pulpa y papel y en la industria textil, sirven como estabilizadores de blanqueadores peróxido, al quelar a los metales que podrían desactivar al peróxido. En detergentes, son usados como una combinación de agentes quelantes, inhibidores de escamas, y estabilizadores de blanqueadores. Los fosfonatos también tienen uso en medicina para tratar desórdenes asociados a la formación de huesos y metabolismo de calcio. Más aún, sirven como portadores de radionúclidos en tratamientos de cáncer a los huesos (ver EDTMP de samario-153).
En 1998, el consumo mundial de fosfonatos fue de 56000 toneladas - 40000 toneladas en USA, 15000 toneladas en Europa y menos de 800 toneladas en Japón. La demanda de fosfonatos ha crecido a un ritmo firme anual del 3%. Junto con los organosilicatos, los fosfonatos también son utilizados para tratar la muerte súbita causada por Phytophthora ramorum.
Toxicología
[editar]La toxicidad de los fosfonatos a organismos acuáticos es baja. Los valores reportados para LC50 y 48h para peces están entre 0,1 y 1,1 mM. También el factor de bioconcentración por los peces es muy bajo.
Biodegradación
[editar]En la naturaleza, las bacterias juegan un rol principal en la degradación de los fosfonatos.[41] Debido a la presencia de fosfonatos naturales en el ambiente, las bacterias han evolucionado la habilidad de metabolizar fosfonatos como fuente de nutrientes. Algunas bacterias usan fosfonatos como una fuente de fósforo para su crecimiento. Los aminofosfonatos también pueden ser usados como fuente de nitrógeno del suelo por algunas bacterias. Los polifosfonatos usados en la industria difieren significativamente de los fosfonatos naturales, tales como el ácido 2-aminoetilfosfónico, porque son más grandes, portan una mayor carga negativa, y están complejados con los metales. Los ensayos de biodegradación con lodo de plantas de tratamiento de alcantarillado municipal con HEDP y NTMP no mostró señales de degradación. Una investigación del HEDP, NTMP, EDTMP, y DTPMP en ensayos de biodegradación estándar también ha fallado en identificar cualquier biodegradación. Sin embargo, se ha observado en algunos ensayos que, debido a la alta relación entre el lodo y el fosfonato, existe remoción de la sustancia de ensayo de la solución observada como pérdida de DOC. Este factor era atribuido a la adsorción en vez de la biodegradación. Sin embargo, se ha aislado cepas bacterianas capaces de degradar aminopolifosfonatos y HEDP bajo condiciones P-limitadas de suelos, lagos, desagües, lodo activado y compost.
No se ha observado biodegradación de fosfonatos durante el tratamiento de agua, pero la fotodegradación de los complejos de Fe(III) es rápida. Los aminopolifosfonatos también son rápidamente oxidados en presencia de Mn(II) y oxígeno, y son formados productos estables de la descomposición, en desagües. La falta de información acerca de fosfonatos en el ambiente está asociada a problemas analíticos de su determinación a concentraciones traza en aguas naturales. Los fosfonatos están presentes principalmente como complejos de Ca y Mg en las aguas naturales y, en consecuencia, no afectan la especiación del metal o su transporte.[42]
Los fosfonatos son una de las tres fuentes de ingesta de fosfato en las células biológicas (siendo los otros dos el fosfato inorgánico y el organofosfato)
Véase también
[editar]- Compuestos de organofósforo
- Fosfina - PR3
- Óxido de fosfina - OPR3
- Fosfinito - P(OR)R2
- Fosfonito - P(OR)2R
- Fosfito - P(OR)3
- Fosfinato - OP(OR)R2
- Fosfato - OP(OR)3
- Ácido fosforoso - H3PO3
Referencias
[editar]- ↑ a b Crystal chemistry of inorganic phosphites, J. Loub, Acta Cryst. (1991), B47, 468—473, doi 10.1107/S0108768191002380
- ↑ a b c Svara, J.; Weferling, N.; Hofmann, T. "Phosphorus Compounds, Organic," in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2008. doi 10.1002/14356007.a19_545.pub2.
- ↑ EU Pesticides database. «Potassium phosphonates».
- ↑ Hardy, G.E.S., Barrett, S. & Shearer, B.L. (2001). «The future of phosphite as a fungicide to control the soilborne plant pathogen Phytophthora cinnamomi in natural ecosystems». Australasian Plant Pathology 30: 133–139. doi:10.1071/AP01012.
- ↑ Elser JJ, et al. (2007). «Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems». Ecol Lett 10 (12): 1135–1142. PMID 17922835. doi:10.1111/j.1461-0248.2007.01113.x.
- ↑ van Veen HW (1997). «Phosphate transport in prokaryotes: molecules, mediators and mechanisms». Antonie Van Leeuwenhoek 72 (4): 299-315. PMID 9442271. doi:10.1023/a:1000530927928.
- ↑ a b Metcalf WW, van der Donk WA. (2009). «Biosynthesis of phosphonic and phosphinic acid natural products». Annu Rev Biochem 78: 65-94. PMID 19489722. doi:10.1146/annurev.biochem.78.091707.100215.
- ↑ HORIGUCHI M, KANDATSU M. (1959). «Isolation of 2-aminoethane phosphonic acid from rumen protozoa.». Nature 184: 901–902. doi:10.1038/184901b0.
- ↑ K.-S. Ju • J. R. Doroghazi • W. W. Metcalf (2014). «Genomics-enabled discovery of phosphonate natural products and their biosynthetic pathways». J Ind Microbiol Biotechnol 41: 345–356. doi:10.1007/s10295-013-1375-2.
- ↑ James S. Kittredge, Eugene Roberts (1969). «A Carbon-Phosphorus Bond in Nature». Science 164 (3875): 37-42. doi:10.1126/science.164.3875.37.
- ↑ Juan F. Villarreal-Chiu, John P. Quinn and John W. McGrath (2012). «The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment». Front. Microbiol. 3 (19). doi:10.3389/fmicb.2012.00019.
- ↑ Dyhrman S.T., Benitez-Nelson C.R., Orchard E.D., Haley S.T., Pellechia P.J. «A microbial source of phosphonates in oligotrophic marine systems». Nature Geoscience 2 (10): 696-699. doi:10.1038/ngeo639.
- ↑ W. W. Metcalf et al. (2012). «Synthesis of Methylphosphonic Acid by Marine Microbes: A Source for Methane in the Aerobic Ocean». Science 337 (6098): 1104-1107. doi:10.1126/science.1219875.
- ↑ Moschidis MC (1985) «Phosphonolipids». Prog Lipid Res 23:223–246
- ↑ Hilderbrand RL (1983) «The role of phosphonates in living systems». CRC Press, USA
- ↑ Hendlin D, et al. (1969). «Phosphonomycin, a new antibiotic produced by strains of streptomyces». Science 166 (3901): 122-123. PMID 5809587. doi:10.1126/science.166.3901.122.
- ↑ Baeyer E, Gugel KH, Haegele K, Hagenmaier H, Jessipow S, Koenig WA, Zaehner J (1972) «Stofwechselprodukte von Mikroorganismen 98. Phosphinothricin and phosphinothricyle-alanylalanin». Helv Chim Acta 55:224–239
- ↑ Omura S, Hinotozawa K, Imamura N, Murata M (1984) «The structure of phosalacine, a new herbicidal antibiotic containing phosphinothricin». J Antibiot 37(8):939–940
- ↑ Omura S, Murata M, Hanaki H, Hinotozawa K, Oiwa R, Tanaka H (1984) «Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action». J Antibiot 37(8):829–835
- ↑ Kato H, Nagayama K, Abe H, Kobayashi R, Ishihara E (1991) «Isolation, structure and biological activity of trialaphos». Agric Biol Chem 55:1133–1134
- ↑ Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H (1980) «Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR32863 and FR-33289». J Antibiot 33(1):24–28
- ↑ Park BK, Hirota A, Sakai H (1977) «Structure of plumbemycin A and B, antagonists of L-threonine from Streptomyces plumbeus». Agric Biol Chem 41:573–579
- ↑ Takeuchi M, Nakajima M, Ogita T, Inukai M, Kodama K, Furuya K, Nagaki H, Haneishi T (1989) «Fosfonochlorin, a new antibiotic with spheroplast forming activity». J Antibiot 42(2):198–205
- ↑ Ogita T, Gunji S, Fukawa Y, Terahara A, Kinoshita T, Nagaki H (1983) «The structures of fosfazinomycins A and B». Tetrahedron Lett 24:2283–2286
- ↑ Rapp C, Jung G, Kugler M, Loeffler W (1988) «Rhizocticins— new phosphono-oligopeptides with antifungal activity». Liebigs Annalen der Chemie 7:655–661
- ↑ Takahashi E, Kimura T, Nakamura K, Arahira M, Iida M (1995) «Phosphonothrixin, a novel herbicidal antibiotic produced by Saccharothrix sp. ST-888. I. Taxonomy, fermentation, isolation and biological properties». J Antibiot 48(10):1124–1129
- ↑ Spencer C Peck; Wilfred A van der Donk (2013). «Phosphonate biosynthesis and catabolism: a treasure trove of unusual enzymology». Current Opinion in Chemical Biology 17 (4): 580-588. doi:10.1016/j.cbpa.2013.06.018.
- ↑ a b Modern Phosphonate Chemistry by Philippe Savignac and Bogdan Iorga, CRC Press, Boca Raton, FL, 2003. ISBN 0-8493-1099-7
- ↑ Greenwood, Norman N.; Earnshaw, Alan. (1997), Chemistry of the Elements (2nd ed.), Oxford: Butterworth-Heinemann, ISBN 0-08-037941-9
- ↑ Daniella Vincze, Péter Ábrányi-Balogh, Péter Bagi, György Keglevich (2019). «A Mechanistic Study on the Tautomerism of H-Phosphonates, H-Phosphinates and Secondary Phosphine Oxides». Molecules 24: 3859. doi:10.3390/molecules24213859.
- ↑ a b c d Kolio D. Troev (2018). Reactivity of P-H Group of Phosphorus Based Compounds. Academic Press. pp. 1-17. ISBN 978-0-12-813834-2.
- ↑ Malowan, John E. (1953). «Diethyl phosphite». Inorganic Syntheses 4: 58-60. doi:10.1002/9780470132357.ch19.
- ↑ A. H. Ford-Moore (1963). "Triethyl Phosphite". Org. Synth.; Coll. Vol. 4: 955.
- ↑ Hoffmann, Friedrich W.; Ess, Richard J.; Usingef, Robert P. (November 1956). «The Transesterification of Trialkyl Phosphites with Aliphatic Alcohols». Journal of the American Chemical Society 78 (22): 5817-5821. doi:10.1021/ja01603a026.
- ↑ Synthesis of phosphorus esters by transesterification mediated by N-heterocyclic carbenes (NHCs), R. Singh, S. P. Nolan, Chem. Commun., 2005, 5456-5458.
- ↑ M. Sekine, K. Okimoto, K. Yamada, T. Hata (1981). «Silyl phosphites. 15. Reactions of silyl phosphites with .alpha.-halo carbonyl compounds. Elucidation of the mechanism of the Perkow reaction and related reactions with confirmed experiments». J. Org. Chem. 46 (10): 2097-2107. doi:10.1021/jo00323a024.
- ↑ A novel Cu(OTf)2 mediated three component high yield synthesis of α-aminophosphonates Abhimanyu S. Paraskar and Arumugam Sudalai Arkivoc 06-1838EP) pp 183-189 2006 Article
- ↑ Kabachnik, Martin I.; T. Ya. Medved (1952). «Новый метод синтеза сс-аминофосфиновых кислот». Doklady Akademii Nauk SSSR 83: 689.
- ↑ Fields, Ellis K. (1952). «The synthesis of esters of substituted amino phosphonic acids». Journal of the American Chemical Society 74 (6): 1528-1531. doi:10.1021/ja01126a054.
- ↑ a b Engel, Robert (2004). «Phosphorus Addition at sp2 Carbon». Organic Reactions 36 (2): 175-248. doi:10.1002/0471264180.or036.02.
- ↑ Huang J, Su Z, Xu Y (November 2005). «The evolution of microbial phosphonate degradative pathways». Journal of Molecular Evolution 61 (5): 682-90. Bibcode:2005JMolE..61..682H. PMID 16245012. doi:10.1007/s00239-004-0349-4.
- ↑ Nowack Bernd (2003). «Environmental chemistry of phosphonates». Water Research 37 (11): 2533-2546. PMID 12753831. doi:10.1016/S0043-1354(03)00079-4.
 Datos: Q422733
Datos: Q422733 Multimedia: Phosphonates / Q422733
Multimedia: Phosphonates / Q422733